

ライブリバント点滴静注350mg
医療用
医療用医薬品:
医師の処方により使用する医薬品
医師の処方により使用する医薬品
- 収載区分
- 銘柄別収載
- 先発・後発情報
- 先発品(後発品なし)
- オーソライズド
ジェネリック - -
- 一般名
- アミバンタマブ(遺伝子組換え)注射液
- 英名(商品名)
- Rybrevant
- 規格
- 350mg7mL1瓶
- 薬価
- 160,014.00
- メーカー名
- ヤンセンファーマ
- 規制区分
- 劇薬
- 長期投与制限
- -
- 標榜薬効
- 抗悪性腫瘍薬〔抗ヒトEGFR/抗ヒトMET完全ヒト型二重特異性モノクローナル抗体〕
- 色
- -
- 識別コード
- -
- [@: メーカーロゴ]
- 添付文書
-
PDF 2026年3月改訂(第6版)
- 告示日
- 2024年11月19日
- 経過措置期限
- -
- 医薬品マスタに反映
- 2024年12月版
- 医薬品マスタ削除予定
- -
- 運転注意
- 情報なし(使用の適否を判断するものではありません)
- ドーピング
- 禁止物質なし(使用の適否を判断するものではありません)
- CP換算
- -
- 長期収載品選定療養
- -
[識別コードの表記 @: メーカーロゴ]
改訂情報
2026年3月31日 DSU No.344 【重要】
【7.用法及び用量に関連する注意】(一部改訂)
ラゼルチニブとの併用投与による静脈血栓塞栓症の発症を抑制するため、当該併用投与開始後4ヵ月間は、アピキサバン1回2.5mgを1日2回経口投与すること。アピキサバンの電子添文を参照して、出血リスクに十分注意すること。ただし、腎不全(クレアチニンクリアランス(CLcr)15mL/min未満)の患者では、アピキサバンは投与できないことから、アミバンタマブ(遺伝子組換え)とラゼルチニブとの併用投与以外の治療選択肢を考慮すること。
【9.2腎機能障害患者】(新設)
〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉
腎不全(CLcr 15mL/min未満)の患者:
アピキサバンは投与できないことから、ラゼルチニブとの併用投与は避け、他の治療選択肢を考慮すること。
2026年3月6日 使用上の注意改訂情報 令和8年3月6日指示分
【7. 用法及び用量に関連する注意】(一部改訂)
ラゼルチニブとの併用投与による静脈血栓塞栓症の発症を抑制するため、当該併用投与開始後4ヵ月間は、アピキサバン1回2.5mgを1日2回経口投与すること。アピキサバンの電子添文を参照して、出血リスクに十分注意すること。ただし、腎不全(クレアチニンクリアランス(CLcr)15mL/min未満)の患者では、アピキサバンは投与できないことから、アミバンタマブ(遺伝子組換え)とラゼルチニブとの併用投与以外の治療選択肢を考慮すること。
【9. 特定の背景を有する患者に関する注意-9.2 腎機能障害患者】(新設)
〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉
腎不全(CLcr 15mL/min未満)の患者
アピキサバンは投与できないことから、ラゼルチニブとの併用投与は避け、他の治療選択肢を考慮すること。
2026年1月27日 DSU No.342 【その他】
【11.1重大な副作用】(一部改訂)
重度の皮膚障害:
発疹、ざ瘡様皮膚炎、皮膚潰瘍等の重度の皮膚障害があらわれることがある。
【11.1重大な副作用】(追記)
体液貯留:
低アルブミン血症、末梢性浮腫、全身性浮腫等の体液貯留があらわれることがある。急激な体重の増加、呼吸困難等の異常が認められた場合には本剤の投与を中止するなど適切な処置を行うこと。
【11.2その他の副作用】(削除)
2026年1月27日 DSU No.342 【その他】
【8.重要な基本的注意】(追記)
本剤の使用にあたっては、アミバンタマブ(遺伝子組換え)・ボルヒアルロニダーゼアルファ(遺伝子組換え)配合皮下注製剤との取り違えに注意すること。
2025年7月8日 DSU No.337 【その他】
【5.効能又は効果に関連する注意】(追記)
〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉
EGFRチロシンキナーゼ阻害剤による治療後に増悪した患者に対してカルボプラチン及びペメトレキセドナトリウムと併用する場合は、臨床試験に組み入れられた患者の前治療歴等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。
【6.用法及び用量】(一部改訂)
EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発の非小細胞肺癌にはA法、EGFR遺伝子変異(エクソン20挿入変異を除く)陽性の切除不能な進行・再発の非小細胞肺癌にはA法又はB法を使用する。
A法:カルボプラチン及びペメトレキセドナトリウムとの併用において、3週間を1サイクルとし、通常、成人にはアミバンタマブ(遺伝子組換え)として以下の用法及び用量で点滴静注する。なお、患者の状態により適宜減量する。
B法:ラゼルチニブメシル酸塩との併用において、4週間を1サイクルとし、通常、成人にはアミバンタマブ(遺伝子組換え)として以下の用法及び用量で点滴静注する。なお、患者の状態により適宜減量する。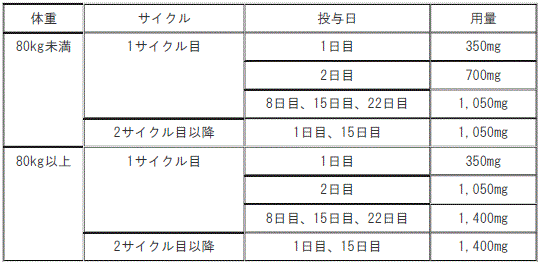
【7.用法及び用量に関連する注意】(追記)
EGFRチロシンキナーゼ阻害剤による治療歴のないEGFR遺伝子変異(エクソン20挿入変異を除く)陽性の切除不能な進行・再発の非小細胞肺癌に対するA法の有効性及び安全性は確立していない。
【7.用法及び用量に関連する注意】(追記)
本剤、ラゼルチニブ、カルボプラチン及びペメトレキセドナトリウムの併用投与は行わないこと。
【14.適用上の注意】(一部改訂)
[薬剤投与時の注意]
輸液ポンプ及び投与セットを用いて、点滴静注により調製後の本剤を投与する。また、投与セットは、滅菌されたパイロジェンフリー(エンドトキシンフリー)の低蛋白結合性のポリエーテルスルホン製、ナイロン製又はポリスルホン製のインラインフィルター(孔径0.2μm又は0.22μm)を備えたポリウレタン、ポリブタジエン、ポリ塩化ビニル、ポリプロピレン又はポリエチレン製を用いること。
【14.適用上の注意】(一部改訂)
調製後の本剤は、室内光下にて室温のもと、溶液の調製開始後10時間以内に投与を完了すること。
効能効果
1). EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発非小細胞肺癌。
2). EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌。
(効能又は効果に関連する注意)
5.1. 〈EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発非小細胞肺癌〉*十分な経験を有する病理医又は検査施設における検査により、EGFR遺伝子エクソン20挿入変異が確認された患者に投与すること(検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること)。
5.2. 〈EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発非小細胞肺癌〉臨床試験に組み入れられた患者の遺伝子変異の種類等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと〔17.1.1参照〕。
5.3. 〈EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発非小細胞肺癌〉本剤の術前・術後補助療法としての有効性及び安全性は確立していない。
5.4. 〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉*十分な経験を有する病理医又は検査施設における検査により、EGFR遺伝子変異(エクソン20挿入変異を除く)が確認された患者に投与すること(検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること)。
5.5. 〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉EGFRチロシンキナーゼ阻害剤による治療後に増悪した患者に対してカルボプラチン及びペメトレキセドナトリウムと併用する場合は、臨床試験に組み入れられた患者の前治療歴等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと〔17.1.3参照〕。
5.6. 〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉本剤の術前・術後補助療法としての有効性及び安全性は確立していない。
*)承認された体外診断用医薬品又は医療機器に関する情報については、次のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html。
用法用量
EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発の非小細胞肺癌にはA法、EGFR遺伝子変異(エクソン20挿入変異を除く)陽性の切除不能な進行・再発の非小細胞肺癌にはA法又はB法を使用する。
A法:カルボプラチン及びペメトレキセドナトリウムとの併用において、3週間を1サイクルとし、通常、成人にはアミバンタマブ(遺伝子組換え)として次の用法及び用量で点滴静注する。なお、患者の状態により適宜減量する。
1). 体重80kg未満:
①. 1サイクル目:1日目350mg、2日目1050mg、8日目・15日目1400mg。
②. 2サイクル目:1日目1400mg。
③. 3サイクル目以降:1日目1750mg。
2). 体重80kg以上:
①. 1サイクル目:1日目350mg、2日目1400mg、8日目・15日目1750mg。
②. 2サイクル目:1日目1750mg。
③. 3サイクル目以降:1日目2100mg。
B法:ラゼルチニブメシル酸塩との併用において、4週間を1サイクルとし、通常、成人にはアミバンタマブ(遺伝子組換え)として以下の用法及び用量で点滴静注する。なお、患者の状態により適宜減量する。
1). 体重80kg未満:
①. 1サイクル目:1日目350mg、2日目700mg、8日目・15日目・22日目1050mg。
②. 2サイクル目以降:1日目・15日目1050mg。
2). 体重80kg以上:
①. 1サイクル目:1日目350mg、2日目1050mg、8日目・15日目・22日目1400mg。
②. 2サイクル目以降:1日目・15日目1400mg。
(用法及び用量に関連する注意)
7.1. 本剤投与によるinfusion reactionを軽減させるため、本剤投与前に、1サイクル目の第1日目及び第2日目は、副腎皮質ホルモン剤、抗ヒスタミン剤及び解熱鎮痛剤を投与し、必要に応じてH2受容体拮抗剤や制吐剤を投与し、投与前に、1サイクル目の第8日目以降は、抗ヒスタミン剤及び解熱鎮痛剤を投与し、必要に応じて副腎皮質ホルモン剤、H2受容体拮抗剤や制吐剤を投与すること〔11.1.1参照〕。
7.2. 調製後の希釈液を次の速度で投与すること。
[本剤の投与量及び投与速度(カルボプラチン及びペメトレキセドナトリウムとの併用の場合)]
1). 体重80kg未満:
①. 1サイクル目:
a. 1日目:投与量350mg/250mL、投与速度(投与開始時)50mL/時、投与速度(投与開始2時間後)75mL/時。
b. 2日目:投与量1050mg/250mL、投与速度(投与開始時)33mL/時、投与速度(投与開始2時間後)50mL/時。
c. 8日目:投与量1400mg/250mL、投与速度65mL/時。
d. 15日目:投与量1400mg/250mL、投与速度85mL/時。
②. 2サイクル目:1日目:投与量1400mg/250mL、投与速度125mL/時。
③. 3サイクル目以降:1日目:投与量1750mg/250mL、投与速度125mL/時。
2). 体重80kg以上:
①. 1サイクル目:
a. 1日目:投与量350mg/250mL、投与速度(投与開始時)50mL/時、投与速度(投与開始2時間後)75mL/時。
b. 2日目:投与量1400mg/250mL、投与速度(投与開始時)25mL/時、投与速度(投与開始2時間後)50mL/時。
c. 8日目:投与量1750mg/250mL、投与速度65mL/時。
d. 15日目:投与量1750mg/250mL、投与速度85mL/時。
②. 2サイクル目:1日目:投与量1750mg/250mL、投与速度125mL/時。
③. 3サイクル目以降:1日目:投与量2100mg/250mL、投与速度125mL/時。
Infusion reactionが認められない場合は、投与開始2時間後に投与速度を上げることができる。
[本剤の投与量及び投与速度(ラゼルチニブとの併用の場合)]
1). 体重80kg未満:
①. 1サイクル目:
a. 1日目:投与量350mg/250mL、投与速度(投与開始時)50mL/時、投与速度(投与開始2時間後)75mL/時。
b. 2日目:投与量700mg/250mL、投与速度(投与開始時)50mL/時、投与速度(投与開始2時間後)75mL/時。
c. 8日目:投与量1050mg/250mL、投与速度85mL/時。
d. 15日目、22日目:投与量1050mg/250mL、投与速度125mL/時。
②. 2サイクル目以降:1日目、15日目:投与量1050mg/250mL、投与速度125mL/時。
2). 体重80kg以上:
①. 1サイクル目:
a. 1日目:投与量350mg/250mL、投与速度(投与開始時)50mL/時、投与速度(投与開始2時間後)75mL/時。
b. 2日目:投与量1050mg/250mL、投与速度(投与開始時)35mL/時、投与速度(投与開始2時間後)50mL/時。
c. 8日目:投与量1400mg/250mL、投与速度65mL/時。
d. 15日目:投与量1400mg/250mL、投与速度85mL/時。
e. 22日目:投与量1400mg/250mL、投与速度125mL/時。
②. 2サイクル目以降:1日目、15日目:投与量1400mg/250mL、投与速度125mL/時。
Infusion reactionが認められない場合は、投与開始2時間後に投与速度を上げることができる。
7.3. ラゼルチニブとの併用投与による静脈血栓塞栓症の発症を抑制するため、当該併用投与開始後4ヵ月間は、アピキサバン1回2.5mgを1日2回経口投与すること(アピキサバンの電子添文を参照して、出血リスクに十分注意すること)。ただし、腎不全(クレアチニンクリアランス
7.4. 本剤投与により副作用が発現した場合には、次を参考に本剤を減量、中断、休薬又は中止すること。
[副作用発現時に本剤を減量する場合の投与量]
1). 副作用発現時の投与量1050mg:1段階減量700mg、2段階減量350mg、3段階減量は中止。
2). 副作用発現時の投与量1400mg:1段階減量1050mg、2段階減量700mg、3段階減量は中止。
3). 副作用発現時の投与量1750mg:1段階減量1400mg、2段階減量1050mg、3段階減量は中止。
4). 副作用発現時の投与量2100mg:1段階減量1750mg、2段階減量1400mg、3段階減量は中止。
[副作用発現時の本剤の処置]
1). Infusion reaction:
①. Grade1のInfusion reaction及びGrade2のInfusion reaction:a.投与を中断する、b.症状が回復した場合、発現時の50%の投与速度で再開する、c.再開後の30分間にinfusion reactionの症状が認められない場合、中断時の投与速度まで上げることができる(その後の2時間にinfusion reactionの症状が認められない場合、同日に予定されていた最高速度まで上げることができる)。d.Grade2のinfusion reactionによる投与中断・再開後にGrade2のinfusion reactionが再発した場合、同日における投与は中止を検討する。
②. Grade3のInfusion reaction:a.同日における投与を中止する、b.次回以降の投与可否は患者の状態に応じて判断し、投与速度はGrade2の場合を参考に患者の状態に応じて判断する、c.Grade3のinfusion reactionが再発した場合、投与を中止する。
③. Grade4のInfusion reaction:投与を中止する。
2). 間質性肺疾患:
①. 間質性肺疾患疑い:休薬する。
②. 間質性肺疾患確定:投与を中止する。
3). 静脈血栓塞栓症(ラゼルチニブとの併用時):
①. 静脈血栓塞栓症(ラゼルチニブとの併用時):臨床的に不安定な事象が発現した場合(例:呼吸不全、心機能障害)、発現した事象が臨床的に安定するまで休薬する。
②. 静脈血栓塞栓症(ラゼルチニブとの併用時):抗凝固剤による治療中に静脈血栓塞栓症再発した場合、投与を中止する。
4). 皮膚障害又は爪障害:
①. Grade1の皮膚障害又はGrade1の爪障害:2週間後に改善が認められない場合、減量を検討する。
②. Grade2の皮膚障害又はGrade2の爪障害:a.ラゼルチニブとの併用時は、減量を検討する、Grade2の皮膚障害又はGrade2の爪障害:b.2週間後に改善が認められない場合、減量を検討する。
③. Grade3の皮膚障害又はGrade3の爪障害:a.Grade2以下に回復するまで休薬し、減量して投与を再開する、Grade3の皮膚障害又はGrade3の爪障害:b.ラゼルチニブとの併用時は休薬し、週1回の観察を行う(2週間以内にGrade2以下に回復した場合は減量を検討した上で投与を再開し、2週間以内にGrade2以下に回復しない場合は投与を中止する)。
④. Grade4の皮膚障害又はGrade4の爪障害、重度水疱性皮膚障害又は重度剥脱性皮膚障害:投与を中止する。
5). その他の副作用:
①. Grade2の副作用:a.休薬を検討する(1週間より後に改善した場合、減量して投与を再開することを検討する)、Grade2の副作用:b.ラゼルチニブとの併用時は、休薬又は減量を検討する(28日以内に改善した場合は同じ用量又は減量して投与を再開することを検討し、28日より後に改善した場合は減量して投与を再開することを検討する)。
②. Grade3の副作用:a.Grade1以下又はベースラインに回復するまで休薬する、b.1週間以内に回復した場合、同じ用量で投与を再開する、c.1週間より後に回復した場合、減量して投与を再開する、d.4週間以内に回復しない場合、投与の中止を検討する。
③. Grade4の副作用:原則として投与を中止する。
GradeはNCI-CTCAE v5.0に準じる。
7.5. EGFR-TKI治療歴のないエクソン20挿入変異を除くEGFR変異陽性の切除不能な進行・再発の非小細胞肺癌に対するA法の有効性及び安全性は確立していない(EGFR-TKI:EGFRチロシンキナーゼ阻害剤)。
7.6. 本剤、ラゼルチニブ、カルボプラチン及びペメトレキセドナトリウムの併用投与は行わないこと。
警告・禁忌・相互作用・その他の注意
(警告)
1.1. 本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
1.2. 本剤の投与により間質性肺疾患があらわれ、死亡に至った症例が報告されているので、初期症状(呼吸困難、咳嗽、発熱等)の確認及び定期的な胸部画像検査の実施等、観察を十分に行い、異常が認められた場合は本剤の投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。また、特に治療初期は入院又はそれに準ずる管理の下で、間質性肺疾患等の重篤な副作用発現に関する観察を十分に行うこと〔8.1、9.1.1、11.1.2参照〕。
1.3. 本剤投与開始前に、胸部CT検査及び問診を実施し、間質性肺疾患の合併又は既往歴の有無を確認した上で、投与の可否を慎重に判断すること〔9.1.1参照〕。
1.4. ラゼルチニブとの併用投与により、深部静脈血栓症及び肺塞栓症を含む静脈血栓塞栓症があらわれ、死亡に至った症例が報告されているので、静脈血栓塞栓症の既往歴の有無等を確認した上で、投与の可否を慎重に判断すること(また、本剤投与中は患者の状態を十分に観察し、下肢疼痛・下肢浮腫、突然の呼吸困難、息切れ、胸痛等の静脈血栓塞栓症が疑われる徴候や症状の発現に注意すること)〔7.3、8.3、9.1.2、11.1.4参照〕。
(禁忌)
本剤の成分に対し過敏症の既往歴のある患者。
(重要な基本的注意)
8.1. 間質性肺疾患があらわれることがあるので、初期症状(呼吸困難、咳嗽、発熱等)の確認及び定期的な胸部画像検査の実施等、観察を十分に行い、必要に応じて、動脈血酸素分圧(PaO2)、動脈血酸素飽和度(SpO2)、肺胞気動脈血酸素分圧較差(A-aDO2)、肺拡散能力(DLCO)等の検査を行うこと。また、患者に対して、間質性肺疾患の初期症状があらわれた場合には、速やかに医療機関を受診するよう指導すること〔1.2、9.1.1、11.1.2参照〕。
8.2. 重度の皮膚障害があらわれることがあるので、必要に応じて皮膚科を受診するよう患者に指導すること〔11.1.3参照〕。
8.3. ラゼルチニブとの併用により静脈血栓塞栓症の発現頻度が増加する傾向が認められているので、初期症状(下肢疼痛・下肢浮腫、突然の呼吸困難、息切れ、胸痛等)の確認及び定期的な凝固能検査の実施等、観察を十分に行うこと。また、患者に対して、静脈血栓塞栓症の初期症状があらわれた場合には、速やかに医療機関を受診するよう指導すること〔1.4、7.3、9.1.2、11.1.4参照〕。
8.4. 本剤の使用にあたっては、アミバンタマブ(遺伝子組換え)・ボルヒアルロニダーゼ アルファ(遺伝子組換え)配合皮下注製剤との取り違えに注意すること。
(特定の背景を有する患者に関する注意)
(合併症・既往歴等のある患者)
9.1.1. 間質性肺疾患のある患者又はその既往歴のある患者:間質性肺疾患が悪化又は再発するおそれがある〔1.2、1.3、8.1、11.1.2参照〕。
9.1.2. 静脈血栓塞栓症のある患者又はその既往歴のある患者:静脈血栓塞栓症が悪化又は再発するおそれがある〔1.4、7.3、8.3、11.1.4参照〕。
(腎機能障害患者)
9.2.1. 〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉腎不全(CLcr15mL/min未満)の患者:アピキサバンは投与できないことから、ラゼルチニブとの併用投与は避け、他の治療選択肢を考慮すること〔7.3参照〕。
(生殖能を有する者)
妊娠する可能性のある女性:妊娠する可能性のある女性には、本剤投与中及び最終投与後3ヵ月間において避妊する必要性及び適切な避妊法について説明すること〔9.5妊婦の項参照〕。
(妊婦)
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること(本剤を用いた生殖発生毒性試験は実施されていないが、類薬のEGFR又はMET阻害剤を投与した動物試験では、胚発生障害・胎仔発生障害、胚致死及び流産の発現率の上昇が認められた)〔9.4生殖能を有する者の項参照〕。
(授乳婦)
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること(本剤のヒト母乳中への移行に関するデータはないが、ヒトIgGは母乳中に移行することが知られている)。
(小児等)
小児等を対象とした臨床試験は実施していない。
(高齢者)
ラゼルチニブとの併用投与については、投与の可否を慎重に判断すること(本剤とラゼルチニブを併用した臨床試験において、65歳未満の患者と比較して65歳以上の患者で死亡に至った有害事象、重篤な有害事象及び投与中止に至った有害事象の発現割合が高い傾向が認められている)。
(適用上の注意)
14.1. 薬剤調製時の注意
14.1.1. 本剤が無色~微黄色であることを確認する(変色又は微粒子が認められた場合は使用しないこと)。
14.1.2. 輸液バッグは、ポリ塩化ビニル、ポリプロピレン、ポリエチレン又はポリオレフィン混合物製を用いること。
14.1.3. 希釈液には250mLの5%ブドウ糖注射液又は0.9%生理食塩液を用いる(輸液バッグに加える本剤と同量の希釈液(本剤1バイアルにつき7mL)を抜き取り廃棄する)。
14.1.4. 各バイアルから本剤7mLを抜き取り、輸液バッグに混和する。
14.1.5. 添加後は穏やかに混和し、振盪しないこと。
14.2. 薬剤投与時の注意
14.2.1. 輸液ポンプ及び投与セットを用いて、点滴静注により調製後の本剤を投与する。また、投与セットは、滅菌されたパイロジェンフリー(エンドトキシンフリー)の低蛋白結合性のポリエーテルスルホン製、ナイロン製又はポリスルホン製のインラインフィルター(孔径0.2μm又は0.22μm)を備える、投与セットは、ポリウレタン、ポリブタジエン、ポリ塩化ビニル、ポリプロピレン又はポリエチレン製を用いること。
14.2.2. 投与前に投与セットを希釈液(5%ブドウ糖溶液又は0.9%生理食塩液)で満たすこと。
14.2.3. 投与前に調製後の本剤を目視検査する(変色又は微粒子が認められた場合は使用しないこと)。
14.2.4. 調製後の本剤は、室内光下にて室温のもと、溶液の調製開始後10時間以内に投与を完了すること。
14.2.5. 他の薬剤<5%ブドウ糖溶液又は0.9%生理食塩液を除く>と同じ静注ラインを用いた同時投与は行わないこと。
14.2.6. 本剤のバイアルは1回使い切りである。未使用残液については適切に廃棄すること。
(その他の注意)
15.1. 臨床使用に基づく情報
臨床試験において、本剤に対する抗体産生が報告されている。
(取扱い上の注意)
20.1. 外箱開封後は遮光して保存すること。
20.2. 凍結させないこと。
(保管上の注意)
2~8℃保存。
副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1. 重大な副作用
11.1.1. Infusion reaction(57.4%):悪寒、悪心、呼吸困難、潮紅、胸部不快感、嘔吐等のinfusion reactionがあらわれることがある(多くの場合は、初回投与時に認められたが、2回目以降の投与時にも認められている)〔7.1参照〕。
11.1.2. 間質性肺疾患:肺臓炎(1.6%)、間質性肺疾患(0.9%)があらわれることがあるので、異常が認められた場合には本剤の投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと〔1.2、8.1、9.1.1参照〕。
11.1.3. 重度の皮膚障害:発疹(15.8%*)、ざ瘡様皮膚炎(6.6%*)、皮膚潰瘍(頻度不明*)等の重度皮膚障害があらわれることがある〔8.2参照〕。
11.1.4. 静脈血栓塞栓症
1). 〈化学療法と併用投与〉静脈血栓塞栓症:肺塞栓症(4.6%※)、深部静脈血栓症(4.0%※)等の静脈血栓塞栓症があらわれることがある〔1.4、7.3、8.3、9.1.2参照〕。
2). 〈ラゼルチニブと併用投与〉静脈血栓塞栓症:肺塞栓症(7.8%※※)、深部静脈血栓症(5.5%※※)等の静脈血栓塞栓症があらわれることがある〔1.4、7.3、8.3、9.1.2参照〕。
11.1.5. 動脈血栓塞栓症:本剤とラゼルチニブとの併用において、心筋梗塞(0.5%※※)等の動脈血栓塞栓症があらわれることがある。
11.1.6. 体液貯留:低アルブミン血症(33.2%)、末梢性浮腫(25.6%)、全身性浮腫(4.4%)等の体液貯留があらわれることがあるので、急激な体重増加、呼吸困難等の異常が認められた場合には本剤の投与を中止するなど適切な処置を行うこと。
*)NCI-CTCAEのGrade3以上の副作用頻度。
※)本剤を化学療法と併用投与した臨床試験(NSC3001試験、NSC3002試験)における発現頻度。
※※)本剤をラゼルチニブと併用投与した臨床試験(NSC3003試験)における発現頻度。
11.2. その他の副作用
1). 感染症及び寄生虫症:(10%以上)爪囲炎(56.0%)、(10%未満1%以上)結膜炎。
2). 血液及びリンパ系障害:(10%以上)血小板減少症、(10%未満1%以上)好中球減少症、白血球減少症。
3). 代謝及び栄養障害:(10%以上)食欲減退、(10%未満1%以上)低カルシウム血症、低カリウム血症、低マグネシウム血症。
4). 神経系障害:(10%未満1%以上)浮動性めまい。
5). 眼障害:(10%未満1%以上)ドライアイ、眼瞼炎、角膜炎、霧視、結膜充血、眼そう痒症、睫毛成長、(1%未満)眼充血、視力障害、上強膜炎、非感染性結膜炎、視力低下、(頻度不明)眼障害、ぶどう膜炎、角膜刺激。
6). 心臓障害:(1%未満)洞性頻脈、動悸、頻脈。
7). 胃腸障害:(10%以上)口内炎(36.5%)、下痢、便秘、悪心、(10%未満1%以上)嘔吐、腹痛、痔核。
8). 皮膚及び皮下組織障害:(10%以上)発疹(66.8%)、ざ瘡様皮膚炎(29.2%)、皮膚乾燥、皮膚そう痒症、(10%未満1%以上)爪毒性、湿疹、(1%未満)乾皮症、皮膚剥脱。
9). 筋骨格系及び結合組織障害:(10%未満1%以上)筋肉痛。
10). 一般・全身障害及び投与部位の状態:(10%以上)無力症、疲労、(10%未満1%以上)末梢腫脹、発熱。
11). 臨床検査:(10%以上)ALT増加(23.6%)、AST増加、(10%未満1%以上)血中ALP増加。
薬物動態
16.1 血中濃度
16.1.1 反復投与
国際共同第I相試験(61186372EDI1001試験)で、切除不能な進行・再発の非小細胞肺癌患者(薬物動態解析対象20例(体重80kg未満:15例、体重80kg以上:5例)、日本人4例を含む)に、本剤注1)をカルボプラチン及びペメトレキセドナトリウムと併用して点滴静注したときの血清中濃度推移及び薬物動態パラメータは次のとおりであった。
また、母集団薬物動態解析に基づく本剤の終末相半減期の幾何平均値(変動係数%)は13.7日(31.9%)と推定された。
注1)3週間を1サイクルとし、体重別に次の用法及び用量で点滴静注した。
体重80kg未満の場合:1サイクル目の1日目に350mg、2日目に1,050mg、8日目、15日目、2サイクル目の1日目に1,400mg、3サイクル目以降は1日目に1,750mg
体重80kg以上の場合:1サイクル目の1日目に350mg、2日目に1,400mg、8日目、15日目、2サイクル目の1日目に1,750mg、3サイクル目以降は1日目に2,100mg
図 血清中アミバンタマブ濃度推移
平均値及び標準偏差
<<図省略>>
表 薬物動態パラメータ
<<表省略>>
化学療法歴のあるEGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌患者(薬物動態解析対象108例(体重80kg未満:93例、体重80kg以上:15例))に、本剤注2)をラゼルチニブと併用して点滴静注したときの80kg未満の患者における薬物動態パラメータは次のとおりであった。また、①80kg未満及び②80kg以上の患者における第2及び4サイクルの投与前濃度は、それぞれ①295±94.4及び140±57.4、②285±43.9及び163±43.0μg/mLであった。
表 薬物動態パラメータ
<<表省略>>
注2)4週間を1サイクルとし、体重別に次の用法及び用量で点滴静注した。
体重80kg未満の場合:1サイクル目の1日目に350mg、2日目に700mg、8日目、15日目、22日目に1,050mg、2サイクル目以降は1日目、15日目に1,050mg
体重80kg以上の場合:1サイクル目の1日目に350mg、2日目に1,050mg、8日目、15日目、22日目に1,400mg、2サイクル目以降は1日目、15日目に1,400mg
臨床成績
17.1 有効性及び安全性に関する試験
〈EGFR遺伝子エクソン20挿入変異陽性の切除不能な進行・再発の非小細胞肺癌〉
17.1.1 国際共同第III相試験(NSC3001試験)
化学療法歴のないEGFR遺伝子エクソン20挿入変異注1)陽性の切除不能な進行・再発の非小細胞肺癌注2)患者308例(日本人34例含む)を対象に、本剤注3)、カルボプラチン注4)及びペメトレキセドナトリウム注5)の併用投与(ACP)と、カルボプラチン注4)及びペメトレキセドナトリウム注5)の併用投与(CP)とを比較する無作為化非盲検試験を実施した。
主要評価項目である盲検下独立中央判定による無増悪生存期間[中央値(95%信頼区間)]は、ACP群で11.37ヵ月(9.79~13.70ヵ月)及びCP群で6.70ヵ月(5.59~7.33ヵ月)であった[ハザード比:0.395、95%信頼区間:0.296~0.528、p<0.0001(層別ログランク検定)、2023年5月3日カットオフ]。
図 無増悪生存期間のKaplan-Meier曲線[NSC3001試験]
<<図省略>>
注1)各国又は地域の認定検査機関における検査でEGFR遺伝子エクソン20挿入変異陽性であることが確認された患者が対象とされた。組入れ後に実施された中央検査の結果が得られた患者において検出された変異は、EGFR遺伝子エクソン20のC-helix又はLoopfollowing C-helix領域(D761~C775)に1つ以上のアミノ酸が挿入した変異であった。
注2)非扁平上皮癌を有することが組織学的又は細胞学的に確認された患者が組み入れられた。
注3)3週間を1サイクルとし、体重別に次の用法及び用量で点滴静注した。
体重80kg未満の場合:1サイクル目の1日目に350mg、2日目に1,050mg、8日目、15日目、2サイクル目の1日目に1,400mg、3サイクル目以降は1日目に1,750mg
体重80kg以上の場合:1サイクル目の1日目に350mg、2日目に1,400mg、8日目、15日目、2サイクル目の1日目に1,750mg、3サイクル目以降は1日目に2,100mg
注4)AUC5mg・min/mL相当量を3週間間隔で4回点滴静注した。
注5)500mg/m2を3週間間隔で点滴静注した。
ACP群151例(日本人19例含む)中150例(99.3%)に副作用が認められた。主な副作用は、発疹98例(64.9%)、爪囲炎84例(55.6%)、Infusion reaction62例(41.1%)、ざ瘡様皮膚炎52例(34.4%)、口内炎51例(33.8%)、低アルブミン血症50例(33.1%)、ALT増加38例(25.2%)、末梢性浮腫36例(23.8%)、AST増加36例(23.8%)等であった。[5.2参照]
〈EGFR遺伝子変異陽性の切除不能な進行・再発の非小細胞肺癌〉
17.1.2 国際共同第III相試験(NSC3003試験)
化学療法歴のないEGFR遺伝子変異陽性注1)の切除不能な進行・再発の非小細胞肺癌患者1,074例(日本人78例含む)を対象に、本剤注2)とラゼルチニブ注3)との併用投与(Ami/Laz)と、オシメルチニブ(Osi)注4)投与を比較する無作為化比較試験を実施した。
主要評価項目である盲検下独立中央判定による無増悪生存期間[中央値(95%信頼区間)]は、Ami/Laz群で23.72ヵ月(19.12~27.66ヵ月)及びOsi群で16.59ヵ月(14.78~18.46ヵ月)であった[ハザード比:0.70、95%信頼区間:0.58~0.85、p=0.0002(層別ログランク検定)、2023年8月11日カットオフ]。
図 無増悪生存期間のKaplan-Meier曲線[NSC3003試験]
<<図省略>>
注1)EGFR遺伝子活性型変異であるエクソン19の欠失(Ex19del)変異又はエクソン21の変異(L858R)が腫瘍組織検体で確認された患者が組み入れられた。
注2)4週間を1サイクルとし、体重別に次の用法及び用量で点滴静注した。
体重80kg未満の場合:1サイクル目の1日目に350mg、2日目に700mg、8日目、15日目、22日目、2サイクル目以降の1日目、15日目に1,050mg
体重80kg以上の場合:1サイクル目の1日目に350mg、2日目に1,050mg、8日目、15日目、22日目、2サイクル目以降の1日目、15日目に1,400mg
注3)240mgを1日1回経口投与した。
注4)80mgを1日1回経口投与した。
Ami/Laz群421例(日本人29例含む)中413例(98.1%)に副作用が認められた。主な副作用は、発疹302例(71.7%)、Infusion reaction265例(62.9%)、爪囲炎262例(62.2%)、口内炎166例(39.4%)、低アルブミン血症162例(38.5%)であった。
17.1.3 国際共同第III相試験(NSC3002試験)
オシメルチニブ単独投与による治療後に増悪した注1)EGFR遺伝子変異陽性注2)の切除不能な進行・再発の非小細胞肺癌注3)患者657例(日本人50例含む)を対象に、本剤注4)、カルボプラチン注5)及びペメトレキセドナトリウム注6)との併用投与(ACP群)とカルボプラチン注5)及びペメトレキセドナトリウム注6)の併用投与(CP群)を比較する無作為化非盲検試験を実施した。
主要評価項目である盲検下独立中央判定による無増悪生存期間[中央値(95%信頼区間)]は、ACP群で6.28ヵ月(5.55~8.41ヵ月)及びCP群で4.17ヵ月(4.04~4.44ヵ月)であった[ハザード比:0.48、95%信頼区間:0.36~0.64、2023年7月10日カットオフ]。
図 無増悪生存期間のKaplan-Meier曲線[NSC3002試験]
<<図省略>>
注1)切除不能な進行・再発の非小細胞肺癌に対する一次治療又は他のEGFRチロシンキナーゼ阻害剤による一次治療後の二次治療としてオシメルチニブが単独投与され、投与期間中又は投与終了後に疾患進行が認められた患者が対象とされた。非小細胞肺癌に対する周術期治療を受けた場合には、周術期治療の最終投与から12ヵ月以降に再発し、切除不能な進行・再発の非小細胞肺癌に対する一次又は二次治療としてオシメルチニブが単独投与され、投与期間中又は投与終了後に疾患進行が認められた患者が対象とされた。切除不能な進行・再発の非小細胞肺癌に対して他の抗悪性腫瘍剤による治療歴のある患者は対象外とされた。
注2)EGFR遺伝子活性型変異であるエクソン19欠失(Ex19del)変異又はエクソン21の変異(L858R)が腫瘍組織検体又は血液検体で確認された患者が組み入れられた。
注3)非扁平上皮癌を有することが組織学的又は細胞学的に確認された患者が組み入れられた。
注4)3週間を1サイクルとし、体重別に次の用法及び用量で点滴静注した。
体重80kg未満の場合:1サイクル目の1日目に350mg、2日目に1,050mg、8日目、15日目、2サイクル目の1日目に1,400mg、3サイクル目以降は1日目に1,750mg
体重80kg以上の場合:1サイクル目の1日目に350mg、2日目に1,400mg、8日目、15日目、2サイクル目の1日目に1,750mg、3サイクル目以降は1日目に2,100mg
注5)AUC5mg・min/mL相当量を3週間間隔で4回点滴静注した。
注6)500mg/m2を3週間間隔で点滴静注した。
ACP群130例(日本人9例を含む)中128例(98.5%)に副作用が認められた。主な副作用は、Infusion reaction76例(58.5%)、発疹69例(53.1%)、爪囲炎47例(36.2%)、口内炎39例(30.0%)、ざ瘡様皮膚炎28例(21.5%)、悪心26例(20.0%)、末梢性浮腫26例(20.0%)であった。[5.5参照]
薬効薬理
18.1 作用機序
アミバンタマブは、ヒトEGFR及びMETに対する抗原結合部位を有するヒト型免疫グロブリン(Ig)G1二重特異性モノクローナル抗体であり、EGFR及びMETの下流のシグナル伝達経路を阻害することに加えて、抗体依存性細胞傷害(ADCC)活性等を介して、腫瘍増殖抑制作用を示すと考えられている。
18.2 抗腫瘍作用
アミバンタマブは、EGFR遺伝子エクソン20挿入変異を有するヒト非小細胞肺癌(NSCLC)由来DFCI127及びDFCI-58細胞株等に対して増殖抑制作用を示した(in vitro)。
アミバンタマブは、DFCI127細胞株を皮下移植したインターロイキン2受容体γ鎖の部分的欠損を有する非肥満型糖尿病/重症複合型免疫不全マウス、EGFR遺伝子エクソン20挿入変異を有するヒトNSCLC由来YU-1163細胞株、NSCLC患者由来LXFE2478及びYHIM-1029腫瘍組織片、並びにL858R変異及びT790M変異を有するNSCLC由来H1975細胞株をそれぞれ皮下移植したヌードマウス等において腫瘍増殖抑制作用を示した(in vivo)。
よく一緒に見られている薬

医師の処方により使用する医薬品。
特定薬剤管理指導加算等の算定対象となる薬剤。
