

ダラキューロ配合皮下注
医療用
医療用医薬品:
医師の処方により使用する医薬品
医師の処方により使用する医薬品
- 収載区分
- 銘柄別収載
- 先発・後発情報
- 先発品(後発品なし)
- オーソライズド
ジェネリック - -
- 一般名
- ダラツムマブ(遺伝子組換え)・ボルヒアルロニダーゼ アルファ(遺伝子組換え)注射液
- 英名(商品名)
- Darzquro
- 規格
- 15mL1瓶
- 薬価
- 407,109.00
- メーカー名
- ヤンセンファーマ
- 規制区分
- 劇薬
- 長期投与制限
- -
- 標榜薬効
- 抗悪性腫瘍薬〔抗CD38モノクローナル抗体〕
- 色
- -
- 識別コード
- -
- [@: メーカーロゴ]
- 添付文書
-
PDF 2025年11月改訂(第8版)
- 告示日
- 2021年5月18日
- 経過措置期限
- -
- 医薬品マスタに反映
- 2021年6月版
- 医薬品マスタ削除予定
- -
- 運転注意
- 情報なし(使用の適否を判断するものではありません)
- ドーピング
- 禁止物質なし(使用の適否を判断するものではありません)
- CP換算
- -
- 長期収載品選定療養
- -
[識別コードの表記 @: メーカーロゴ]
改訂情報
2026年1月27日 DSU No.342 【その他】
【5.効能又は効果に関連する注意】(追記)
〈高リスクのくすぶり型多発性骨髄腫における進展遅延〉
臨床試験に組み入れられた患者の高リスクの定義等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、診療ガイドライン等の最新の情報を参考に、本剤の投与が適切と判断される患者に使用すること。
【6.用法及び用量】(追記)
〈高リスクのくすぶり型多発性骨髄腫における進展遅延〉
通常、成人には本剤1回15mL(ダラツムマブ(遺伝子組換え)として1,800mg及びボルヒアルロニダーゼアルファ(遺伝子組換え)として30,000単位(2,000単位/mL))を皮下投与する。28日間を1サイクルとし、第1及び2サイクルは1週間間隔で4回(1、8、15、22日目)、第3~6サイクルは2週間間隔で2回(1、15日目)、第7サイクル以降は4週間間隔で1回(1日目)皮下投与する。ただし、投与期間は3年間までとする。
【7.用法及び用量に関連する注意】(削除)
〈効能共通〉
本剤を単独投与した場合の有効性及び安全性は確立していない。
【7.用法及び用量に関連する注意】(追記)
〈多発性骨髄腫〉
本剤を単独投与した場合の有効性及び安全性は確立していない。
【7.用法及び用量に関連する注意】(追記)
〈全身性ALアミロイドーシス〉
本剤を単独投与した場合の有効性及び安全性は確立していない。
【9.8高齢者】(一部改訂)
患者の状態を観察しながら慎重に投与すること。高齢者では一般に生理機能が低下している。ダラツムマブ(遺伝子組換え)点滴静注製剤の臨床試験において、再発又は難治性の多発性骨髄腫患者のうち65歳未満と比較して65歳以上で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎、敗血症であった。造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者において、75歳未満と比較して75歳以上で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎であった。
本剤の臨床試験において、造血幹細胞移植の適応となる未治療の多発性骨髄腫患者、造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者、未治療の全身性ALアミロイドーシス患者及び高リスクのくすぶり型多発性骨髄腫患者において、65歳以上における主な重篤な有害事象は肺炎であった。
【11.2その他の副作用】(一部改訂)
【15.1臨床使用に基づく情報】(一部改訂)
本剤投与によりダラツムマブ(遺伝子組換え)に対する抗体産生が認められた患者の割合は、0.4%(6/1445例)であり、この6例のうち5例においては、ダラツムマブ(遺伝子組換え)に対する中和抗体を認めた。また、ボルヒアルロニダーゼアルファ(遺伝子組換え)に対する抗体産生が認められた患者の割合は、8.1%(117/1445例)であった。
2025年7月8日 DSU No.337 【その他】
【7.用法及び用量に関連する注意】(追記)
〈多発性骨髄腫〉
未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、骨髄抑制、血小板減少又は好中球減少が発現した場合には、下表を参照し本剤の休薬等、適切な処置を行うこと。
本剤の休薬基準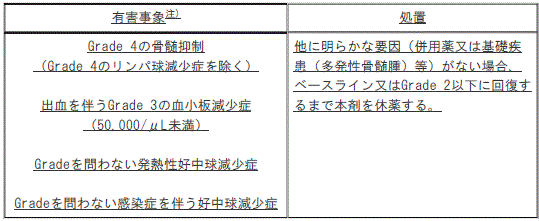
注)GradeはNCI-CTCAE v5.0に準じる。
【7.用法及び用量に関連する注意】(追記)
自家造血幹細胞移植の適応となる未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、レナリドミドの用法及び用量は以下のとおりとすること。
本剤、ボルテゾミブ及びデキサメタゾンとの併用において、レナリドミドとして1日1回25mgを21日間連日経口投与した後、7日間休薬する。これを1サイクルとして投与を繰り返す。
その後は、本剤との併用において、レナリドミドとして1日1回10mgを連日経口投与し、12週間投与後に忍容性が認められる場合には1日1回15mgに増量できる。なお、患者の状態により適宜減量する。
【7.用法及び用量に関連する注意】(追記)
未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、血小板減少又は好中球減少が発現した場合には、下表を参照しレナリドミドの休薬・減量等を考慮すること。
減量する場合のレナリドミドの投与量
(本剤、ボルテゾミブ及びデキサメタゾンとの併用時)
【7.用法及び用量に関連する注意】(追記)
減量する場合のレナリドミドの投与量
(本剤との併用時)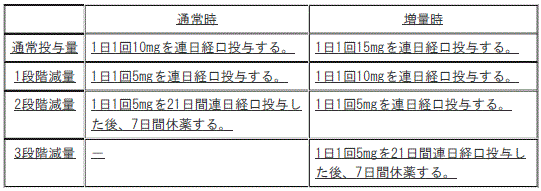
レナリドミドの休薬・減量基準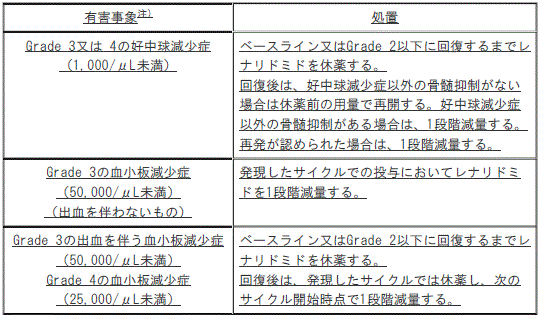
注)GradeはNCI-CTCAE v5.0に準じる。
【9.8高齢者】(一部改訂)
患者の状態を観察しながら慎重に投与すること。高齢者では一般に生理機能が低下している。ダラツムマブ(遺伝子組換え)点滴静注製剤の臨床試験において、再発又は難治性の多発性骨髄腫患者のうち65歳未満と比較して65歳以上で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎、敗血症であった。造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者において、75歳未満と比較して75歳以上で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎であった。
本剤の臨床試験において、造血幹細胞移植の適応となる未治療の多発性骨髄腫患者、造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者、及び未治療の全身性ALアミロイドーシス患者において、65歳以上における主な重篤な有害事象は肺炎であった。
【11.2その他の副作用】(一部改訂)
効能効果
1). 多発性骨髄腫。
2). 全身性ALアミロイドーシス。
3). 高リスクのくすぶり型多発性骨髄腫における進展遅延。
(効能又は効果に関連する注意)
5.1. 〈多発性骨髄腫〉「17.臨床成績」の項及びダラツムマブ(遺伝子組換え)点滴静注製剤の添付文書の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと〔17.1.1-17.1.11参照〕。
5.2. 〈全身性ALアミロイドーシス〉「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと〔17.1.12参照〕。
5.3. 〈高リスクのくすぶり型多発性骨髄腫における進展遅延〉臨床試験に組み入れられた患者の高リスクの定義等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、診療ガイドライン等の最新の情報を参考に、本剤の投与が適切と判断される患者に使用すること〔17.1.13参照〕。
用法用量
〈多発性骨髄腫〉
他の抗悪性腫瘍剤との併用において、通常、成人には本剤1回15mL(ダラツムマブ(遺伝子組換え)として1800mg及びボルヒアルロニダーゼ アルファ(遺伝子組換え)として30000単位(2000単位/mL))を、併用する抗悪性腫瘍剤の投与サイクルを考慮して、次のA法又はB法の投与間隔で皮下投与する。
A法:1週間間隔、2週間間隔及び4週間間隔の順で投与する。
B法:1週間間隔、3週間間隔及び4週間間隔の順で投与する。
〈全身性ALアミロイドーシス〉
他の薬剤との併用において、通常、成人には本剤1回15mL(ダラツムマブ(遺伝子組換え)として1800mg及びボルヒアルロニダーゼ アルファ(遺伝子組換え)として30000単位(2000単位/mL))を皮下投与する。
投与間隔は、1週間間隔、2週間間隔及び4週間間隔の順で投与とする。
〈高リスクのくすぶり型多発性骨髄腫における進展遅延〉
通常、成人には本剤1回15mL(ダラツムマブ(遺伝子組換え)として1800mg及びボルヒアルロニダーゼ アルファ(遺伝子組換え)として30000単位(2000単位/mL))を皮下投与する。28日間を1サイクルとし、第1及び2サイクルは1週間間隔で4回(1、8、15、22日目)、第3~6サイクルは2週間間隔で2回(1、15日目)、第7サイクル以降は4週間間隔で1回(1日目)皮下投与する。ただし、高リスクのくすぶり型多発性骨髄腫の場合、投与期間は3年間までとする。
(用法及び用量に関連する注意)
7.1. 〈効能共通〉本剤投与によるinfusion reactionを軽減させるために、本剤投与開始1~3時間前に副腎皮質ホルモン、解熱鎮痛剤及び抗ヒスタミン剤を投与すること。また、遅発性infusion reactionを軽減させるために、必要に応じて本剤投与後に副腎皮質ホルモン等を投与すること〔11.1.1参照〕。
7.2. 〈効能共通〉Infusion reactionが発現した場合、次のように、本剤の投与中止、投与速度の変更等、適切な処置を行うこと。なお、GradeはNCI-CTCAE v4.0に準じる〔11.1.1参照〕。
・ 〈効能共通〉Grade3のinfusion reactionが3回発現した場合は本剤の投与を中止すること。
・ 〈効能共通〉Grade4のinfusion reactionが発現した場合は本剤の投与を中止すること。
7.3. 〈多発性骨髄腫〉本剤を単独投与した場合の有効性及び安全性は確立していない。
7.4. 〈多発性骨髄腫〉本剤の投与間隔、投与間隔の変更時期、本剤と併用する抗悪性腫瘍剤等について、「17.臨床成績」の項及びダラツムマブ(遺伝子組換え)点滴静注製剤の添付文書の内容を熟知した上で選択すること〔17.1.1-17.1.11参照〕。
7.5. 〈多発性骨髄腫〉ボルテゾミブ及びデキサメタゾン併用、又はボルテゾミブ、メルファラン及びプレドニゾロン併用の場合、併用投与終了後も本剤単独投与を継続すること。
7.6. 〈多発性骨髄腫〉未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、骨髄抑制、血小板減少又は好中球減少が発現した場合には、次を参照し本剤の休薬等、適切な処置を行うこと[本剤の休薬基準:Grade4の骨髄抑制
GradeはNCI-CTCAE v5.0に準じる。
7.7. 〈多発性骨髄腫〉自家造血幹細胞移植の適応となる未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、レナリドミドの用法及び用量は次のとおりとすること。
本剤、ボルテゾミブ及びデキサメタゾンとの併用において、レナリドミドとして1日1回25mgを21日間連日経口投与した後、7日間休薬する。これを1サイクルとして投与を繰り返す。
その後は、本剤との併用において、レナリドミドとして1日1回10mgを連日経口投与し、12週間投与後に忍容性が認められる場合には1日1回15mgに増量できる。なお、患者の状態により適宜減量する。
7.8. 〈多発性骨髄腫〉未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合、血小板減少又は好中球減少が発現した場合には、次を参照しレナリドミドの休薬・減量等を考慮すること。
[減量する場合のレナリドミドの投与量(本剤、ボルテゾミブ及びデキサメタゾンとの併用時)]
1). 〈多発性骨髄腫〉通常投与量:25mg。
2). 〈多発性骨髄腫〉1段階減量:20mg。
3). 〈多発性骨髄腫〉2段階減量:15mg。
4). 〈多発性骨髄腫〉3段階減量:10mg。
5). 〈多発性骨髄腫〉4段階減量:5mg。
6). 〈多発性骨髄腫〉5段階減量:投与中止。
[減量する場合のレナリドミドの投与量(本剤との併用時)]
1). 〈多発性骨髄腫〉通常投与量:(通常時)1日1回10mgを連日経口投与する、(増量時)1日1回15mgを連日経口投与する。
2). 〈多発性骨髄腫〉1段階減量:(通常時)1日1回5mgを連日経口投与する、(増量時)1日1回10mgを連日経口投与する。
3). 〈多発性骨髄腫〉2段階減量:(通常時)1日1回5mgを21日間連日経口投与した後、7日間休薬する、(増量時)1日1回5mgを連日経口投与する。
4). 〈多発性骨髄腫〉3段階減量:(増量時)1日1回5mgを21日間連日経口投与した後、7日間休薬する。
[レナリドミドの休薬・減量基準]
1). 〈多発性骨髄腫〉未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合でGrade3の好中球減少症又はGrade4の好中球減少症(好中球1000/μL未満):ベースライン又はGrade2以下に回復するまでレナリドミドを休薬し、回復後は、好中球減少症以外の骨髄抑制がない場合は休薬前のレナリドミドの用量で再開し、好中球減少症以外の骨髄抑制がある場合は、1段階減量する(再発が認められた場合は、レナリドミドを1段階減量する)。
2). 〈多発性骨髄腫〉未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合でGrade3の血小板減少症<50000/μL未満><出血を伴わないもの>:発現したサイクルでの投与においてレナリドミドを1段階減量する。
3). 〈多発性骨髄腫〉未治療の多発性骨髄腫患者に対して、ボルテゾミブ、レナリドミド及びデキサメタゾンと併用する場合でGrade3の出血を伴う血小板減少症<50000/μL未満>、Grade4の血小板減少症(血小板25000/μL未満):ベースライン又はGrade2以下に回復するまでレナリドミドを休薬し、回復後は、発現したサイクルではレナリドミドを休薬し、次のサイクル開始時点で1段階減量する。
GradeはNCI-CTCAE v5.0に準じる。
7.9. 〈全身性ALアミロイドーシス〉本剤を単独投与した場合の有効性及び安全性は確立していない。
7.10. 〈全身性ALアミロイドーシス〉本剤の投与間隔、投与間隔の変更時期、本剤と併用する薬剤等について、「17.臨床成績」の項の内容を熟知した上で選択すること〔17.1.12参照〕。
警告・禁忌・相互作用・その他の注意
(警告)
本剤の投与は、緊急時に十分対応できる医療施設において、造血器悪性腫瘍又は全身性ALアミロイドーシスの治療に対して十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例のみに行うこと。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分に説明し、同意を得てから投与を開始すること。
(禁忌)
本剤の成分に対し過敏症の既往歴のある患者。
(重要な基本的注意)
8.1. 骨髄抑制があらわれることがあるので、本剤の投与前及び投与中は、定期的に血液検査等を行い、患者の状態を十分に観察すること〔9.1.3、11.1.2参照〕。
8.2. 本剤は、赤血球上に発現しているCD38と結合し、間接クームス試験結果が偽陽性となる可能性があり、当該干渉は本剤最終投与より6ヵ月後まで持続する可能性があるため、本剤投与前に不規則抗体のスクリーニングを含めた一般的な輸血前検査の実施をすること。輸血が予定されている場合は、本剤を介した間接クームス試験への干渉について関係者に周知すること〔12.1参照〕。
8.3. 腫瘍崩壊症候群があらわれることがあるので、血清中電解質濃度及び腎機能検査を行う等、患者の状態を十分に観察すること〔11.1.4参照〕。
8.4. 本剤の投与によりB型肝炎ウイルス再活性化による肝炎があらわれることがあるので、本剤投与に先立って肝炎ウイルス感染の有無を確認し、本剤投与前に適切な処置を行うこと〔9.1.2、11.1.3参照〕。
8.5. 本剤の使用にあたっては、ダラツムマブ(遺伝子組換え)点滴静注製剤との取り違えに注意すること。
(特定の背景を有する患者に関する注意)
(合併症・既往歴等のある患者)
9.1.1. 慢性閉塞性肺疾患若しくは気管支喘息のある患者又はそれらの既往歴のある患者:本剤の投与後処置として気管支拡張剤及び吸入ステロイド剤の投与を考慮すること(本剤投与後に遅発性気管支痙攣を含む気管支痙攣の発現リスクが高くなるおそれがある)。
9.1.2. B型肝炎ウイルスキャリアの患者又はHBs抗原陰性でHBc抗体陽性若しくはHBs抗原陰性でHBs抗体陽性の患者:本剤の投与開始後は継続して肝機能検査や肝炎ウイルスマーカーのモニタリングを行うなど、B型肝炎ウイルス再活性化の徴候や症状の発現に注意すること(本剤の投与によりB型肝炎ウイルス再活性化による肝炎があらわれることがある)〔8.4、11.1.3参照〕。
9.1.3. 体重65kg以下の患者:好中球減少等の骨髄抑制の発現が増加することがある〔8.1、11.1.2参照〕。
(生殖能を有する者)
妊娠可能な女性及びパートナーが妊娠する可能性のある男性:妊娠可能な女性及びパートナーが妊娠する可能性のある男性に対しては、本剤投与中及び本剤投与終了後一定期間は適切な避妊を行うよう指導すること。男性の受胎能に対する影響は検討されておらず不明である〔9.5妊婦の項参照〕。
(妊婦)
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること(本剤を用いた生殖発生毒性試験は実施されていないが、IgG1モノクローナル抗体に胎盤通過性があることが知られている)〔9.4生殖能を有する者の項参照〕。
(授乳婦)
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること(本剤のヒト乳汁中への移行は検討されていないが、ヒトIgGは乳汁中に移行するので、本剤も移行する可能性がある)。
(小児等)
小児等を対象とした臨床試験は実施していない。
(高齢者)
患者の状態を観察しながら慎重に投与すること(高齢者では一般に生理機能が低下している)。ダラツムマブ(遺伝子組換え)点滴静注製剤の臨床試験において、65歳未満と比較して65歳以上の再発又は難治性の多発性骨髄腫患者で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎、敗血症であった。ダラツムマブ(遺伝子組換え)点滴静注製剤の臨床試験において、75歳未満と比較して75歳以上の造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者で重篤な有害事象の発現頻度は高く、主な重篤な有害事象は肺炎であった。
本剤の臨床試験において、造血幹細胞移植の適応となる65歳以上の未治療の多発性骨髄腫患者、造血幹細胞移植の適応とならない65歳以上の未治療の多発性骨髄腫患者、65歳以上の未治療の全身性ALアミロイドーシス患者及び65歳以上の高リスクのくすぶり型多発性骨髄腫患者における主な重篤な有害事象は肺炎であった。
(臨床検査結果に及ぼす影響)
12.1. 本剤は赤血球上のCD38と結合し、抗体スクリーニングや交差試験等の適合性試験に干渉する。本剤による間接クームス試験への干渉を回避するために、ジチオスレイトール(DTT)処理(本剤と赤血球上のCD38との結合を阻害する)を考慮すること。Kell血液型抗原はDTT処理で変性するので、不規則抗体スクリーニングにおいてKell血液型抗原に対する抗体の評価が不能となることに注意すること〔8.2参照〕。
12.2. 本剤はヒトIgGκ型モノクローナル抗体であり、血清中Mタンパクの血清蛋白電気泳動法及び血清免疫固定法の結果に干渉する可能性があり、IgGκ型多発性骨髄腫細胞を有する患者における完全奏効(CR)の評価及びCRからの再発の評価に影響を及ぼす可能性があるため注意すること。
(適用上の注意)
14.1. 薬剤調製時の注意
14.1.1. 本剤の投与には、ポリプロピレン又はポリエチレンのシリンジとステンレス鋼製の注射針を用いること(翼状針で投与する場合は、ポリプロピレン、ポリエチレン又はポリ塩化ビニル(PVC)のチューブ、コネクター等を用いること)。
14.1.2. 本剤は、無菌環境下において、調製すること。
14.1.3. 本剤を冷蔵庫から取り出し、15~30℃に戻しておくこと(未穿刺バイアルは、室温及び室内光下で最長24時間保管ができる)。
14.1.4. 注射針の詰まりを避けるために、投与直前に皮下注射針又は皮下投与セットをシリンジに取り付ける。
14.1.5. 薬液入りシリンジを直ちに使用しない場合は、本剤調製後、室温及び室内光下で7時間まで保存できる(本剤調製後直ちに冷蔵庫に保存した場合は、最長24時間保存の後、室温及び室内光下で7時間まで保存できる)。
14.2. 薬剤投与時の注意
14.2.1. 本剤投与前に粒子や変色の有無を目視で確認すること(不透明粒子や変色又は異物が認められた場合は使用しないこと)。
14.2.2. 臍から左又は右に約7.5cmの腹部皮下に、本剤15mLを約3~5分かけて投与する(他の部位への投与はデータが得られていないため行わない)。
14.2.3. 同一部位への反復注射は行わないこと。
14.2.4. 皮膚の発赤・挫傷・圧痛・硬結又は瘢痕がある部位には注射しないこと。
14.2.5. 患者が痛みを感じた場合は、注射速度を減速又は注射を中断する(減速しても痛みが軽減しない場合は、残りを左右逆側の腹部に投与することができる)。
14.2.6. 本剤投与中は、同一部位に他剤を皮下投与しないこと。
14.2.7. 本剤は1回使い切りである(未使用残液については適切に廃棄すること)。
(その他の注意)
15.1. 臨床使用に基づく情報
本剤投与によりダラツムマブ(遺伝子組換え)に対する抗体産生が認められた患者の割合は、0.4%(6/1445例)であり、この6例のうち5例においては、ダラツムマブ(遺伝子組換え)に対する中和抗体を認めた。また、ボルヒアルロニダーゼ アルファ(遺伝子組換え)に対する抗体産生が認められた患者の割合は、8.1%(117/1445例)であった。
(取扱い上の注意)
20.1. 激しく振盪しないこと。
20.2. 外箱開封後は遮光して保存すること。
(保管上の注意)
2~8℃保存。
副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1. 重大な副作用
11.1.1. Infusion reaction:アナフィラキシー、鼻閉、咳嗽、悪寒、眼障害(脈絡膜滲出、急性近視、急性閉塞隅角緑内障等)、気管支痙攣、低酸素症、呼吸困難等のinfusion reaction(32.7%)があらわれることがあり、多くの場合は、初回投与時に発現が認められたが、2回目以降の投与時にも認められているので、異常が認められた場合は、本剤の投与を中断又は中止し適切な処置を行うとともに、症状が回復するまで患者の状態を十分に観察すること。重度infusion reactionが認められた場合、本剤の投与中止等の適切な処置を行うこと〔7.1、7.2参照〕。
11.1.2. 骨髄抑制:好中球減少(19.6%)、血小板減少(15.1%)、リンパ球減少(7.3%)及び発熱性好中球減少症(1.1%)等の骨髄抑制があらわれることがある〔8.1、9.1.3参照〕。
11.1.3. 感染症:肺炎(8.7%)や敗血症(1.1%)等の重篤な感染症や、B型肝炎ウイルス再活性化があらわれることがある〔8.4、9.1.2参照〕。
11.1.4. 腫瘍崩壊症候群(頻度不明):異常が認められた場合には適切な処置(生理食塩液、高尿酸血症治療剤等の投与、透析等)を行うとともに、症状が回復するまで患者の状態を十分に観察すること〔8.3参照〕。
11.1.5. 間質性肺疾患(0.2%):異常が認められた場合には、本剤の投与を中止し、必要に応じて、胸部CT、血清マーカー等の検査を実施するとともに、適切な処置を行うこと。
11.2. その他の副作用
1). 感染症及び寄生虫症:(10%以上)上気道感染、(5%未満)気管支炎、尿路感染、COVID-19、サイトメガロウイルス感染。
2). 血液及びリンパ系障害:(10%未満5%以上)貧血、白血球減少。
3). 免疫系障害:(5%未満)低γグロブリン血症。
4). 代謝及び栄養障害:(5%未満)食欲減退、低カリウム血症、低カルシウム血症、脱水、高血糖。
5). 精神障害:(5%未満)不眠症。
6). 神経系障害:(5%未満)末梢性ニューロパチー、頭痛、浮動性めまい、錯感覚、失神。
7). 心臓障害:(5%未満)心房細動。
8). 血管障害:(5%未満)高血圧。
9). 呼吸器、胸郭及び縦隔障害:(5%未満)咳嗽、呼吸困難、(頻度不明)肺水腫。
10). 胃腸障害:(10%未満5%以上)下痢、(5%未満)悪心、便秘、嘔吐、腹痛。
11). 皮膚及び皮下組織障害:(5%未満)発疹、皮膚そう痒症。
12). 筋骨格系及び結合組織障害:(5%未満)筋骨格痛、筋痙縮、関節痛。
13). 一般・全身障害及び投与部位の状態:(10%未満5%以上)注射部位反応、疲労、発熱、無力症、(5%未満)注射部位紅斑、悪寒、末梢性浮腫。
薬物動態
16.1 血中濃度
16.1.1 MMY1008試験(国内試験、単剤療法)
日本人の再発又は難治性の多発性骨髄腫患者6例に、本剤15mLを1週間隔で8週、続いて2週間隔で16週、それ以降は4週間隔で反復皮下投与した。初回投与後の血清中ダラツムマブ濃度推移を添付文書の図1に示す。また、初回投与及び1週間隔での最終(8回目)投与後の薬物動態パラメータを表1に示す。
図1 日本人の再発又は難治性の多発性骨髄腫患者6例に本剤を初回投与したときの血清中ダラツムマブ濃度推移(平均値±標準偏差)
C:サイクル、D:日
<<図省略>>
表1 日本人の再発又は難治性の多発性骨髄腫患者6例における本剤初回投与時及び1週間隔での最終(8回目、第50日)投与時のダラツムマブの薬物動態パラメータ
<<表省略>>
16.1.2 MMY3012試験(国際共同試験、単剤療法)
再発又は難治性の多発性骨髄腫患者259例に、本剤15mLを1週間隔で8週、続いて2週間隔で16週、それ以降は4週間隔で反復皮下投与した。平均血清中ダラツムマブ濃度は、初回投与から3日後に124μg/mL、1週間隔での最終(8回目)投与から1週間後(2週間隔投与への移行日)の投与前に582μg/mL、2週間隔での初回投与から3日目に738μg/mL、2週間隔での最終投与から2週間後(4週間隔投与への移行日)の投与前に555μg/mL、4週間隔投与への移行から約5ヵ月後の投与前に297μg/mLであった。
16.1.3 MMY2040試験(国際共同試験、ボルテゾミブ、メルファラン及びプレドニゾロン又はprednisone※との併用療法)
未治療の多発性骨髄腫患者67例に、本剤15mLをボルテゾミブ、メルファラン及びプレドニゾロン又はprednisone※との併用療法にて1週間隔で6週、続いて3週間隔で48週、それ以降は4週間隔で反復皮下投与した。平均血清中ダラツムマブ濃度は、初回投与から3日後に99μg/mL、1週間隔での最終(6回目)投与から1週間後(3週間隔投与への移行日)の投与前に482μg/mL、3週間隔での初回投与から3日目に612μg/mL、3週間隔での3回目投与の投与前に392μg/mLであった。
※:国内未承認
16.1.4 AMY3001試験(国際共同第III相試験)
未治療の全身性ALアミロイドーシス患者381例を対象に、本剤をボルテゾミブ、シクロホスファミド水和物及びデキサメタゾンとの併用療法にて1週間隔で8週、続いて2週間隔で16週、それ以降は4週間隔で反復皮下投与した。平均血清中ダラツムマブ濃度は、初回投与から4日後に149μg/mL、2週間隔での初回投与(9週目)の投与前及び投与4日後に597μg/mL及び708μg/mL、4週間隔での初回投与(25週目)の投与前に478μg/mL、4週間隔投与への移行後から約5ヵ月後の投与前に273μg/mLであった。
臨床成績
17.1 有効性及び安全性に関する試験
〈多発性骨髄腫〉
17.1.1 MMY3012試験(国際共同第III相臨床試験)
プロテアソーム阻害剤及び免疫調節薬を含む3レジメン以上の前治療歴を有する、又はプロテアソーム阻害剤及び免疫調節薬に治療抵抗性の再発又は難治性の多発性骨髄腫患者522例を対象に、ダラツムマブ(遺伝子組換え)点滴静注製剤単独療法に対する本剤単独療法の非劣性を確認するランダム化非盲検群間比較試験を実施した。主要評価項目の一つである中央判定による奏効率は、本剤群では41.1%(95%信頼区間:35.1~47.3%)(108/263例)、ダラツムマブ(遺伝子組換え)点滴静注製剤群では37.1%(95%信頼区間:31.2~43.3%)(96/259例)であり、奏効率の比は1.11(95%信頼区間:0.89~1.37)であった。また、もう一つの主要評価項目である最高血清中トラフ濃度(第3サイクルの第1日目投与前)(平均値±標準偏差)は、本剤群では593±306μg/mL、ダラツムマブ(遺伝子組換え)点滴静注製剤群では522±226μg/mLであり、最高血清中トラフ濃度の幾何平均比は107.93%(90%信頼区間:95.74~121.67%)であった。以上より、ダラツムマブ(遺伝子組換え)点滴静注製剤群に対する本剤群の非劣性が検証された(2019年1月8日クリニカルカットオフ)。
注1)本剤の用法及び用量:28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で皮下投与した。
注2)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注3)非劣性の判定基準:①奏効率について、ダラツムマブ(遺伝子組換え)点滴静注製剤群に対する本剤群の奏効率の比の95%信頼区間の下限値が60%以上であり、かつ②最高血清中トラフ濃度について、ダラツムマブ(遺伝子組換え)点滴静注製剤群に対する本剤群の幾何平均比の90%信頼区間の下限値が80%以上の場合に、ダラツムマブ(遺伝子組換え)点滴静注製剤群に対する本剤群の非劣性が検証されたとすることとされた。
本剤群260例中134例(51.5%)に副作用が認められた。主な副作用は、infusion reaction 68例(26.2%)、好中球減少症32例(12.3%)、血小板減少症24例(9.2%)、上気道感染21例(8.1%)、貧血21例(8.1%)等であった(2019年7月8日クリニカルカットオフ)。[5.1、7.4参照]
17.1.2 MMY2040試験(国際共同第II相臨床試験)
多発性骨髄腫患者132例(日本人患者4例を含む)を対象に、ボルテゾミブ、メルファラン及びプレドニゾロン又はprednisone※との併用療法(MPB療法)に本剤を上乗せした本剤/MPB療法、並びにレナリドミド及びデキサメタゾンの併用療法(Ld療法)に本剤を上乗せした本剤/Ld療法の有効性及び安全性を検討する非盲検非対照試験を実施した。中央判定による奏効率は、本剤/MPB群では88.1%(90%信頼区間:79.5~93.9%)(59/67例)、本剤/Ld群では90.8%(90%信頼区間:82.6~95.9%)(59/65例)であった。
注1)本剤の用法及び用量:本剤/MPB群では、1~9サイクルまでは42日間を1サイクル、10サイクル以降は28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~6週目)、3週間間隔(7~54週目)及び4週間間隔(55週目以降)で皮下投与した。本剤/Ld療法では、28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で皮下投与した。
注2)ボルテゾミブの用法及び用量:21日間を1サイクルとし、1.3mg/m2を第1~2サイクルでは週2回(1、4、8及び11日目)、第3~18サイクルでは週1回(1及び8日目)皮下投与又は静脈内投与した。なお、症状に応じ適宜減量した。
注3)メルファランの用法及び用量:42日間を1サイクルとし、9サイクルまで9mg/m2を1、2、3及び4日目に経口投与した。なお、症状に応じ適宜減量した。
注4)プレドニゾロン又はprednisone※の用法及び用量:42日間を1サイクルとし、9サイクルまで60mg/m2を1、2、3及び4日目に経口投与した。なお、症状に応じ適宜減量した。
注5)レナリドミドの用法及び用量:28日間を1サイクルとし、CrCL>60mL/minの被験者には25mgを、CrCL30~60mL/minの被験者には10mgを1日1回、21日間経口投与した。なお、症状に応じ適宜減量した。
注6)デキサメタゾンの用法及び用量:28日間を1サイクルとし、40mgを1、8、15及び22日目に静脈内又は経口投与した。なお、症状に応じ適宜減量した。
※:国内未承認
本剤が投与された安全性評価対象例132例中93例(70.5%)に副作用が認められた。主な副作用は、Infusion reaction 33例(25.0%)、好中球減少33例(25.0%)、血小板減少32例(24.2%)、発熱23例(17.4%)、リンパ球減少19例(14.4%)等であった。[5.1、7.4参照]
17.1.3 MMY3019試験(国際共同第III相臨床試験)
造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者395例(日本人患者22例を含む)を対象に、ボルテゾミブ、レナリドミド及びデキサメタゾンの併用療法(BLd療法)とBLd療法に本剤を上乗せしたDBLd療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である微小残存病変(MRD)陰性率は、DBLd群では53.3%(105/197例)、BLd群では35.4%(70/198例)であり、DBLd群で統計学的に有意な改善を示した[オッズ比:2.07、95%信頼区間:1.38~3.10、p=0.0004(フィッシャーの正確検定)、(2021年4月8日クリニカルカットオフ)]。副次評価項目である無増悪生存期間の中央値は、いずれの群も未到達であった[ハザード比:0.61、95%信頼区間:0.42~0.90、p=0.0104(層別Log-rank検定、有意水準両側0.0145)、(2022年9月8日クリニカルカットオフ、中間解析)]。
無増悪生存期間のKaplan-Meier曲線[MMY3019試験]
<<図省略>>
DBLd群:本剤+ボルテゾミブ+レナリドミド+デキサメタゾン、BLd群:ボルテゾミブ+レナリドミド+デキサメタゾン
注1)本剤の用法及び用量:1~8サイクルまでは21日間を1サイクル、9サイクル以降は28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~6週目)、3週間間隔(7~24週目)及び4週間間隔(25週目以降)で皮下投与した。
注2)ボルテゾミブの用法及び用量:21日間を1サイクルとし、8サイクルまで1.3mg/m2を週2回(1、4、8及び11日目)皮下投与した。なお、症状に応じ適宜減量した。
注3)レナリドミドの用法及び用量:1~8サイクルまでは21日間を1サイクル、9サイクル以降は28日間を1サイクルとし、CrCL≧60mL/minの被験者には25mgを、CrCL30~59mL/minの被験者には10mgを1日1回、8サイクルまでは14日間、9サイクル以降は21日間経口投与した。なお、症状に応じ適宜減量した。
注4)デキサメタゾンの用法及び用量:1~8サイクルまでは21日間を1サイクル、9サイクル以降は28日間を1サイクルとし、1~8サイクルまでは20mgを1、2、4、5、8、9、11及び12日目(75歳超又はBMIが18.5未満の場合、1、4、8及び11日目でもよいとされた)に経口投与した。9サイクル以降は40mg(75歳超又はBMIが18.5未満の場合、20mgでもよいとされた)を1、8、15及び22日目に経口投与した。なお、症状に応じ適宜減量した。
注5)微小残存病変(MRD)陰性率:ランダム化後、疾患進行前又は新規抗骨髄腫療法開始前に実施した骨髄穿刺により、完全奏効(CR)以上の奏効が得られ、微小残存病変(MRD)陰性(10の-5乗未満)となった被験者の割合。
DBLd群197例中152例(77.2%)に副作用が認められた。主な副作用は、infusion reaction 79例(40.1%)、好中球減少60例(30.5%)、血小板減少47例(23.9%)、貧血31例(15.7%)、下痢28例(14.2%)等であった。(2024年5月7日クリニカルカットオフ)[5.1、7.4参照]
17.1.4 MMY3014試験(海外第III相臨床試験)
造血幹細胞移植の適応となる未治療の多発性骨髄腫患者709例を対象に、ボルテゾミブ、レナリドミド及びデキサメタゾン併用(BLd)療法(BLdによる寛解導入療法、造血幹細胞移植後のBLdによる地固め療法及びレナリドミド単剤による維持療法)とBLd療法に本剤を上乗せしたDBLd療法(DBLdによる寛解導入療法、造血幹細胞移植後のDBLdによる地固め療法及びレナリドミドと本剤の併用による維持療法)を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、いずれの群も未到達であり、DBLd群で統計学的に有意な延長を示した[ハザード比:0.42、95%信頼区間:0.30~0.59、p<0.0001(層別Log-rank検定、有意水準両側0.0126)、(2023年8月1日クリニカルカットオフ、中間解析)]。
無増悪生存期間のKaplan-Meier曲線[MMY3014試験]
<<図省略>>
DBLd群:本剤+ボルテゾミブ+レナリドミド+デキサメタゾン、BLd群:ボルテゾミブ+レナリドミド+デキサメタゾン
注1)本剤の用法及び用量:28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、寛解導入療法期(第1~4サイクル)の第1~2サイクルでは1週間間隔、寛解導入療法期(第1~4サイクル)の第3~4サイクル及び地固め療法期(第5~6サイクル)では2週間間隔、維持療法期(第7サイクル以降)では4週間間隔で皮下投与した。12カ月間以上微小残存病変(MRD)陰性(10の-5乗未満)を維持し、かつ維持療法を24カ月以上実施している場合、本剤の投与を中止し、レナリドミド単剤による維持療法を継続した。その後完全奏効(CR)からの再発又は微小残存病変(MRD)陽性が認められた場合は本剤の投与を再開した。
注2)ボルテゾミブの用法及び用量:28日間を1サイクルとし、寛解導入療法期(第1~4サイクル)及び地固め療法期(第5~6サイクル)まで1.3mg/m2を週2回(1、4、8及び11日目)皮下投与した。なお、症状に応じ適宜減量した。
注3)レナリドミドの用法及び用量:28日間を1サイクルとし、寛解導入療法期(第1~4サイクル)及び地固め療法期(第5~6サイクル)はCrCL≧50mL/minの被験者には25mgを、CrCL30~49mL/minの被験者には10mgを1日1回、21日間経口投与した。維持療法期(第7サイクル以降)はCrCL≧50mL/minの被験者には10mgを、CrCL30~49mL/minの被験者には5mgを1日1回、28日間経口投与した。なお、症状に応じ適宜減量した。維持療法期の3サイクル終了後(第10サイクル以降)、忍容可能な場合は15mgに増量可能とした。
注4)デキサメタゾンの用法及び用量:28日間を1サイクルとし、寛解導入療法期(第1~4サイクル)及び地固め療法期(第5~6サイクル)まで40mgを1~4及び9~12日目に経口投与した。
注5)造血幹細胞移植:DBLd群及びBLd群において、寛解導入療法期の後に造血幹細胞を採取し、造血幹細胞移植に伴う大量化学療法を実施した。
DBLd群351例中244例(69.5%)に副作用が認められた。主な副作用は、infusion reaction 100例(28.5%)、好中球減少101例(28.8%)、血小板減少75例(21.4%)、上気道感染36例(10.3%)、無力症28例(8.0%)等であった。(2023年8月1日クリニカルカットオフ)[5.1、7.4参照]
17.1.5 MMY3013試験(海外第III相臨床試験)
レナリドミド及びプロテアソーム阻害剤を含む1レジメン以上の前治療歴を有する再発又は難治性の多発性骨髄腫患者304例を対象に、ポマリドミド及びデキサメタゾンの併用療法(Pd療法)とPd療法に本剤又はダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDPd療法注1)を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DPd群では12.4ヵ月(95%信頼区間:8.34~19.32)、Pd群では6.9ヵ月(95%信頼区間:5.52~9.26)であり、DPd群で統計学的に有意な延長を示した[ハザード比:0.63、95%信頼区間:0.47~0.85、p=0.0018(層別Log-rank検定)]。副次評価項目である全生存期間の中央値は、いずれの群も未到達であり、統計学的に有意な延長は認められていない[ハザード比:0.91、95%信頼区間:0.61~1.35、p=0.6359(層別Log-rank検定)、(2020年7月21日クリニカルカットオフ)]。
注1)MMY3013試験開始時点ではダラツムマブ(遺伝子組換え)点滴静注製剤の投与が規定されていたが、試験実施中に新規に投与開始する患者は本剤を投与するよう変更された。この変更前にダラツムマブ(遺伝子組換え)点滴静注製剤の投与を開始していた患者は9週目以降に本剤投与への切替えが許容されていた。ダラツムマブ(遺伝子組換え)点滴静注製剤の投与例はDPd群の151例中7例であり、そのうち4例は投与期間中に本剤に切り替えた。
無増悪生存期間のKaplan-Meier曲線[MMY3013試験]
<<図省略>>
DPd群:本剤又はダラツムマブ(遺伝子組換え)点滴静注製剤+ポマリドミド+デキサメタゾン、Pd群:ポマリドミド+デキサメタゾン
注2)本剤又はダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)又は点滴静注製剤1回16mg/kgを1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で皮下投与又は点滴静注した。
注3)ポマリドミドの用法及び用量:28日間を1サイクルとし、4mgを1日1回、1~21日目まで経口投与した。なお、症状に応じ適宜減量した。
注4)デキサメタゾンの用法及び用量:28日間を1サイクルとし、75歳未満は40mg、75歳以上は20mgを1週間間隔で静脈内又は経口投与した。なお、症状に応じ適宜減量した。
DPd群(ダラツムマブ(遺伝子組換え)点滴静注製剤投与例を除く)の安全性評価対象例142例中86例(60.6%)に副作用が認められた。主な副作用は、好中球減少43例(30.3%)、Infusion reaction 30例(21.1%)、白血球減少23例(16.2%)、肺炎23例(16.2%)、血小板減少21例(14.8%)等であった。[5.1、7.4参照]
17.1.6 (参考)MM-014試験(国際共同第II相臨床試験)コホートC:点滴静注製剤
レナリドミドを含む1又は2レジメンの前治療歴を有する再発又は難治性の日本人多発性骨髄腫患者18例を対象に、ダラツムマブ(遺伝子組換え)点滴静注製剤、ポマリドミド及びデキサメタゾンの併用療法の有効性及び安全性を検討する非盲検非対照試験を実施した。主要評価項目である奏効率は、83.3%(95%信頼区間:58.6~96.4)であった(2020年8月3日クリニカルカットオフ)。
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、1回16mg/kgを1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注2)ポマリドミドの用法及び用量:28日間を1サイクルとし、4mgを1日1回、1~21日目まで経口投与した。なお、症状に応じ適宜減量した。
注3)デキサメタゾンの用法及び用量:28日間を1サイクルとし、75歳以下は40mg、75歳超は20mgを1週間間隔で経口投与した。なお、症状に応じ適宜減量した。
安全性評価症例において、18例中17例(94.4%)に副作用が認められた。主な副作用は、好中球減少12例(66.7%)、Infusion reaction 5例(27.8%)、白血球減少4例(22.2%)、血小板減少3例(16.7%)であった。[5.1、7.4参照]
17.1.7 (参考)20160275(CANDOR)試験(国際共同第III相臨床試験):点滴静注製剤
1~3レジメンの前治療歴を有する再発又は難治性の多発性骨髄腫患者466例(日本人患者31例を含む)を対象に、カルフィルゾミブ(週2回投与)及びデキサメタゾンの併用療法(Cd療法)とCd療法にダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDCd療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DCd群では未到達、Cd群で15.8ヵ月(95%信頼区間:12.1~推定不能)であり、DCd群で統計学的に有意な延長を示した[ハザード比0.630、95%信頼区間:0.464~0.854、p=0.0014(層別log-rank検定)、2019年7月14日クリニカルカットオフ]。副次評価項目である全生存期間の中央値は、いずれの群も未到達であり、統計学的に有意な延長は認められていない[ハザード比0.745、95%信頼区間:0.491~1.131、p=0.0836(層別log-rank検定)、2019年7月14日クリニカルカットオフ]。
無増悪生存期間のKaplan-Meier曲線[20160275(CANDOR)試験]
DCd群:ダラツムマブ(遺伝子組換え)点滴静注製剤+カルフィルゾミブ+デキサメタゾン、Cd群:カルフィルゾミブ+デキサメタゾン
<<図省略>>
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~8週目、初回のみ2日間に分割して8mg/kgずつ投与)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注2)カルフィルゾミブの用法及び用量(週2回投与):28日間を1サイクルとし、1日1回、1、2、8、9、15、16日目に点滴静注した。投与量は、1サイクル目の1、2日目のみ20mg/m2(体表面積)、それ以降は56mg/m2(体表面積)で点滴静注した。
注3)デキサメタゾンの用法及び用量:28日間を1サイクルとし、20mgを1、2、8、9、15、16日目に、40mgを22日目に静脈内又は経口投与した。デキサメタゾンの投与日がカルフィルゾミブと同日の場合、カルフィルゾミブ投与の4時間~30分前、ダラツムマブ(遺伝子組換え)点滴静注製剤投与の1~3時間前に投与した。
DCd群308例中198例(64.3%)に副作用が認められた。主な副作用は、infusion reaction 127例(41.2%)、血小板減少症65例(21.1%)、貧血41例(13.3%)、上気道感染27例(8.8%)、肺炎26例(8.4%)、疲労23例(7.5%)であった。[5.1、7.4参照]
17.1.8 (参考)MMY3008試験(海外第III相臨床試験):点滴静注製剤
造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者737例を対象に、レナリドミド及びデキサメタゾンの併用療法(Ld療法)とLd療法にダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDLd療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DLd群では未到達、Ld群で31.9ヵ月(95%信頼区間:28.9~推定不能)であり、DLd群で統計学的に有意な延長を示した[ハザード比:0.56、95%信頼区間:0.43~0.73、p<0.0001(層別Log-rank検定)、2018年9月24日クリニカルカットオフ]。
無増悪生存期間のKaplan-Meier曲線[MMY3008試験]
DLd群:ダラツムマブ(遺伝子組換え)点滴静注製剤+レナリドミド+デキサメタゾン、Ld群:レナリドミド+デキサメタゾン
<<図省略>>
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注2)レナリドミドの用法及び用量:28日間を1サイクルとし、CrCL>50mL/minの被験者には25mgを、CrCL30~50mL/minの被験者には10mgを1日1回、21日間経口投与した。なお、症状に応じ適宜減量した。
注3)デキサメタゾンの用法及び用量:28日間を1サイクルとし、40mgを1、8、15及び22日目に静脈内又は経口投与した。なお、症状に応じ適宜減量した。
DLd群364例中308例(84.6%)に副作用が認められた。主な副作用は、infusion reaction 203例(55.8%)、好中球減少96例(26.4%)、疲労70例(19.2%)、呼吸困難50例(13.7%)、貧血49例(13.5%)等であった。[5.1、7.4参照]
17.1.9 (参考)MMY3007試験(国際共同第III相臨床試験):点滴静注製剤
造血幹細胞移植の適応とならない未治療の多発性骨髄腫患者680例(日本人患者24例を含む)を対象に、ボルテゾミブ、メルファラン及びプレドニゾロン又はprednisone※の併用療法(MPB療法)とMPB療法にダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDMPB療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DMPB群では未到達、MPB群で17.9ヵ月(95%信頼区間:16.1~19.8)であり、DMPB群で統計学的に有意な延長を示した[ハザード比:0.51、95%信頼区間:0.39~0.67、p<0.0001(層別Log-rank検定)、2017年6月12日クリニカルカットオフ]。副次評価項目である全生存期間の中央値は、いずれの群も未到達であり、DMPB群で統計学的に有意な延長を示した[ハザード比:0.63、95%信頼区間:0.47~0.83、p=0.0009(非層別Log-rank検定)、(2019年6月24日クリニカルカットオフ)]。
無増悪生存期間のKaplan-Meier曲線[MMY3007試験]
DMPB群:ダラツムマブ(遺伝子組換え)点滴静注製剤+ボルテゾミブ+メルファラン+プレドニゾロン又はprednisone※、MPB群:ボルテゾミブ+メルファラン+プレドニゾロン又はprednisone※
※:国内未承認
<<図省略>>
全生存期間のKaplan-Meier曲線[MMY3007試験]
DMPB群:ダラツムマブ(遺伝子組換え)点滴静注製剤+ボルテゾミブ+メルファラン+プレドニゾロン又はprednisone※、MPB群:ボルテゾミブ+メルファラン+プレドニゾロン又はprednisone※
※:国内未承認
<<図省略>>
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:1~9サイクルまでは42日間を1サイクル、10サイクル以降は28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~6週目)、3週間間隔(7~54週目)及び4週間間隔(55週目以降)で点滴静注した。
注2)ボルテゾミブの用法及び用量:21日間を1サイクルとし、1.3mg/m2を第1~2サイクルでは週2回(1、4、8及び11日目)、第3~18サイクルでは週1回(1及び8日目)皮下投与又は静脈内投与した。なお、症状に応じ適宜減量した。
注3)メルファランの用法及び用量:42日間を1サイクルとし、9サイクルまで9mg/m2を1、2、3及び4日目に経口投与した。なお、症状に応じ適宜減量した。
注4)プレドニゾロン又はprednisone※の用法及び用量:42日間を1サイクルとし、9サイクルまで60mg/m2を1、2、3及び4日目に経口投与した。なお、症状に応じ適宜減量した。
※:国内未承認
DMPB群333例中193例(58.0%)に副作用が認められた。主な副作用は、infusion reaction 103例(30.9%)、好中球減少71例(21.3%)、血小板減少63例(18.9%)、貧血28例(8.4%)、呼吸困難24例(7.2%)等であった。(2017年6月12日クリニカルカットオフ)[5.1、7.4参照]
17.1.10 (参考)MMY3003試験(国際共同第III相臨床試験):点滴静注製剤
1レジメン以上の前治療歴を有する再発又は難治性の多発性骨髄腫患者569例(日本人患者36例を含む)を対象に、レナリドミド及びデキサメタゾンの併用療法(Ld療法)とLd療法にダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDLd療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DLd群では未到達、Ld群で18.4ヵ月(95%信頼区間:13.9~推定不能)であり、DLd群で統計学的に有意な延長を示した[ハザード比:0.37、95%信頼区間:0.27~0.52、p<0.0001(層別Log-rank検定)]。副次評価項目である全生存期間の中央値は、DLd群では未到達、Ld群で20.3ヵ月であり、統計学的に有意な延長は認められていない[ハザード比:0.64、95%信頼区間:0.40~1.01、p=0.0534(非層別Log-rank検定)、2016年3月7日クリニカルカットオフ]。
無増悪生存期間のKaplan-Meier曲線[MMY3003試験]
DLd群:ダラツムマブ(遺伝子組換え)点滴静注製剤+レナリドミド+デキサメタゾン、Ld群:レナリドミド+デキサメタゾン
<<図省略>>
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注2)レナリドミドの用法及び用量:28日間を1サイクルとし、CrCL>60mL/minの被験者には25mgを、CrCL30~60mL/minの被験者には10mgを1日1回、21日間経口投与した。なお、症状に応じ適宜減量した。
注3)デキサメタゾンの用法及び用量:28日間を1サイクルとし、40mgを1、8、15及び22日目に静脈内又は経口投与した。なお、症状に応じ適宜減量した。
注4)レナリドミドに対して治療抵抗性を示す又は忍容性が不良の患者は除外した。
DLd群283例中215例(76.0%)に副作用が認められた。主な副作用は、infusion reaction 158例(55.8%)、好中球減少43例(15.2%)、上気道感染43例(15.2%)、疲労35例(12.4%)、咳嗽34例(12.0%)等であった。[5.1、7.4参照]
17.1.11 (参考)MMY3004試験(海外第III相臨床試験):点滴静注製剤
1レジメン以上の前治療歴を有する再発又は難治性の多発性骨髄腫患者498例を対象に、ボルテゾミブ及びデキサメタゾンの併用療法(Bd療法)とBd療法にダラツムマブ(遺伝子組換え)点滴静注製剤を上乗せしたDBd療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である無増悪生存期間の中央値は、DBd群では未到達、Bd群で7.2ヵ月(95%信頼区間:6.2~7.9)であり、DBd群で統計学的に有意な延長を示した[ハザード比:0.39、95%信頼区間:0.28~0.53、p<0.0001(層別Log-rank検定)]。副次評価項目である全生存期間の中央値は、いずれの群も未到達であり、統計学的に有意な延長は認められていない[ハザード比:0.77、95%信頼区間:0.47~1.26、p=0.2975(非層別Log-rank検定)、(2016年1月11日クリニカルカットオフ)]。
無増悪生存期間のKaplan-Meier曲線[MMY3004試験]
DBd群:ダラツムマブ(遺伝子組換え)点滴静注製剤+ボルテゾミブ+デキサメタゾン、Bd群:ボルテゾミブ+デキサメタゾン
<<図省略>>
注1)ダラツムマブ(遺伝子組換え)点滴静注製剤の用法及び用量:1~8サイクルまでは21日間を1サイクル、9サイクル以降は28日間を1サイクルとし、1回16mg/kgを、1週間間隔(1~9週目)、3週間間隔(10~24週目)及び4週間間隔(25週目以降)で点滴静注した。
注2)ボルテゾミブの用法及び用量:21日間を1サイクルとし、1.3mg/m2を週2回(1、4、8及び11日目)8サイクルまで静脈内投与又は皮下投与した。なお、症状に応じ適宜減量した。
注3)デキサメタゾンの用法及び用量:21日間を1サイクルとし、8サイクルまで20mgを1、2、4、5、8、9、11及び12日目に静脈内又は経口投与した。なお、症状に応じ適宜減量した。
注4)ボルテゾミブ、イキサゾミブ若しくはカルフィルゾミブに対して治療抵抗性を示す又はボルテゾミブに対し忍容性が不良の患者は除外した。
DBd群243例中182例(74.9%)に副作用が認められた。主な副作用は、infusion reaction 120例(49.4%)、血小板減少73例(30.0%)、呼吸困難34例(14.0%)、咳嗽30例(12.3%)、疲労27例(11.1%)等であった。[5.1、7.4参照]
〈全身性ALアミロイドーシス〉
17.1.12 AMY3001試験(国際共同第III相臨床試験)
未治療の全身性ALアミロイドーシス患者388例を対象に、シクロホスファミド水和物、ボルテゾミブ及びデキサメタゾンの併用療法(CyBorD療法)と本剤を上乗せしたDCyBorD療法を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である血液学的完全奏効(CR)率は、DCyBorD群では53.3%(95%信頼区間:46.1~60.5)(104/195例)、CyBorD群では18.1%(95%信頼区間:13.0~24.3)(35/193例)であり、DCyBorD群で統計学的に有意な改善を示した[オッズ比:5.13、95%信頼区間:3.22~8.16、p<0.0001(層別Cochran-Mantel-Haenszel検定)、有意水準:0.04999、2020年2月14日クリニカルカットオフ]。
注1)本剤の用法及び用量:28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で24サイクルまで皮下投与した。
注2)シクロホスファミド水和物の用法及び用量:28日間を1サイクルとし、300mg/m2(無水物換算)を1週間間隔で6サイクルまで経口又は静脈内投与した。なお、症状に応じ適宜減量した。
注3)ボルテゾミブの用法及び用量:28日間を1サイクルとし、1.3mg/m2を1週間間隔で6サイクルまで皮下投与した。なお、症状に応じ適宜減量した。
注4)デキサメタゾンの用法及び用量:28日間を1サイクルとし、40mgを1週間間隔で6サイクルまで経口投与した。なお、症状に応じ適宜減量した。
DCyBorD群193例中110例(57.0%)に副作用が認められた。主な副作用は、infusion reaction 53例(27.5%)、リンパ球減少26例(13.5%)、貧血21例(10.9%)、上気道感染21例(10.9%)、注射部位反応21例(10.9%)、疲労18例(9.3%)、血小板減少症16例(8.3%)等であった。DCyBorD群の72.5%がベースライン時に全身性ALアミロイドーシスに関連する心臓障害を有していた。心臓障害関連の有害事象は、心不全8.3%、動悸5.7%、心房細動5.7%であり、重篤又は致死的な心臓障害関連の有害事象は心不全6.2%、心停止3.6%、心房細動2.1%であった。重篤又は致死的な心臓障害を発現した患者はベースライン時に全身性ALアミロイドーシスに関連する心臓障害を有していた。なお、臨床試験ではMayo Clinic Cardiac Staging Systemに基づく心臓病期stageIIIb(NT-proBNP>8,500pg/mL)、NYHA分類クラスIIIB又はIVの患者は除外された。[5.2、7.10参照]
〈高リスクのくすぶり型多発性骨髄腫における進展遅延〉
17.1.13 SMM3001試験(国際共同第III相臨床試験)
高リスク注1)のくすぶり型多発性骨髄腫注2)患者390例(日本人患者28例を含む)を対象に、本剤単独投与と積極的経過観察を比較するランダム化非盲検群間比較試験を実施した。主要評価項目である多発性骨髄腫への進展又は死亡までの期間注3)の中央値は、本剤群では未到達、積極的経過観察群では41.46ヵ月(95%信頼区間:26.41~53.32)であり、本剤群で統計学的に有意な延長を示した[ハザード比:0.49、95%信頼区間:0.36~0.67、p<0.0001(層別Log-rank検定)、有意水準(両側)0.05、2024年5月1日クリニカルカットオフ]。
多発性骨髄腫への進展又は死亡までの期間注3)のKaplan-Meier曲線[SMM3001試験]
注3)無作為化した日からIMWG2014基準に基づく多発性骨髄腫への進展が初めて記録された日又は死亡日のいずれか早い方までの期間と定義。
<<図省略>>
注1)次の基準を1つ以上満たす場合、高リスクと定義。
(1)血清Mタンパク濃度30g/L以上
(2)免疫グロブリン(Ig)A型のくすぶり型多発性骨髄腫
(3)IgA、IgM及びIgGのうち2種類のuninvolved(非腫瘍由来)Igの減少を伴う免疫不全
(4)involved/uninvolved FLC(血清遊離軽鎖)比が8以上100未満
(5)クローナルな骨髄形質細胞(BMPC)が50%超かつ60%未満で測定可能病変を有する
注2)International Myeloma Working Group(IMWG)2014基準に基づくくすぶり型多発性骨髄腫と診断された患者。
注3)無作為化した日からIMWG2014基準に基づく多発性骨髄腫への進展が初めて記録された日又は死亡日のいずれか早い方までの期間と定義。
注4)本剤の用法及び用量:28日間を1サイクルとし、ダラツムマブとして1回1,800mg(ボルヒアルロニダーゼ アルファ30,000単位を含む)を、1週間間隔(1~8週目)、2週間間隔(9~24週目)及び4週間間隔(25週目以降)で39サイクル(最長36ヵ月)まで皮下投与した。
本剤群193例中138例(71.5%)に副作用が認められた。主な副作用は、infusion reaction95例(49.2%)、上気道感染47例(24.4%)、疲労29例(15.0%)、注射部位紅斑28例(14.5%)、咳嗽21例(10.9%)等であった。[5.3参照]
薬効薬理
18.1 作用機序
本剤は、ダラツムマブ及びボルヒアルロニダーゼ アルファを含有する配合剤である。ダラツムマブは、ヒトCD38に結合し、補体依存性細胞傷害(CDC)活性、抗体依存性細胞傷害(ADCC)活性、抗体依存性細胞貪食(ADCP)活性等により、腫瘍の増殖を抑制すると考えられている。ボルヒアルロニダーゼ アルファは、結合組織におけるヒアルロン酸を加水分解する酵素である。
本剤は、ボルヒアルロニダーゼ アルファによりヒアルロン酸が加水分解され、皮下組織における浸透性が増加することで、拡散吸収されたダラツムマブが腫瘍の増殖を抑制すると考えられている。
よく一緒に見られている薬

医師の処方により使用する医薬品。
特定薬剤管理指導加算等の算定対象となる薬剤。
