

ケサンラ点滴静注液350mg
医療用
医療用医薬品:
医師の処方により使用する医薬品
医師の処方により使用する医薬品
医薬品コード(YJコード):1190409A1020
- 収載区分
- 銘柄別収載
- 先発・後発情報
- 先発品(後発品なし)
- オーソライズド
ジェネリック - -
- 一般名
- ドナネマブ(遺伝子組換え)注射液
- 英名(商品名)
- Kisunla
- 規格
- 350mg20mL1瓶
- 薬価
- 66,948.00
- メーカー名
- 日本イーライリリー
- 規制区分
- 劇薬
- 長期投与制限
- -
- 標榜薬効
- アルツハイマー型認知症治療薬〔ヒト化抗N3pGアミロイドβモノクローナル抗体〕
- 色
- -
- 識別コード
- -
- [@: メーカーロゴ]
- 添付文書
-
PDF 2026年6月改訂(第5版)
- 告示日
- 2024年11月19日
- 経過措置期限
- -
- 医薬品マスタに反映
- 2024年12月版
- 医薬品マスタ削除予定
- -
- 運転注意
- 情報なし(使用の適否を判断するものではありません)
- ドーピング
- 禁止物質なし(使用の適否を判断するものではありません)
- CP換算
- -
- 長期収載品選定療養
- -
[識別コードの表記 @: メーカーロゴ]
改訂情報
2026年6月5日 使用上の注意改訂情報 令和8年6月5日指示分
【8. 重要な基本的注意】(一部改訂)
ARIAの発現は、本剤投与開始から24週間以内に多く、重篤なARIAの発現は12週間以内に多いことから、この期間は特に注意深く患者の状態を観察すること。当該期間にかかわらず、ARIAを示唆する症状(頭痛、錯乱、悪心、嘔吐、ふらつき、めまい、振戦、視覚障害、言語障害、認知機能の悪化、意識変容、発作等)が認められた場合には、速やかに医療機関を受診するように患者及び家族・介護者に指導すること。速やかに臨床評価を行い、ARIAの発現が疑われる場合はMRI検査を実施すること。
【8. 重要な基本的注意】(一部改訂)
ARIAを示唆する症状が認められない場合であっても、本剤2回目の投与前、3回目の投与前、4回目の投与前、及び7回目の投与前、並びにそれ以降も定期的にMRI検査を実施し、ARIAの有無を確認すること。
2026年3月31日 DSU No.344 【その他】
【10.2併用注意】(一部改訂)
2025年9月24日 DSU No.339 【その他】
【5.効能又は効果に関連する注意】(一部改訂)
「17.臨床成績」の項の内容を熟知し、第Ⅲ相試験で用いられた診断基準、組み入れられた患者の臨床症状スコアの範囲、試験結果等を十分に理解した上で本剤投与の適否を判断すること。
【5.効能又は効果に関連する注意】(一部改訂)
フロルタウシピル(18F)を用いたPET検査の結果から軽度以上のタウ蓄積が認められると判断できない患者に対する有効性及び安全性は確立していない。本剤の投与に先立ち、アミロイドβ病理に関する検査結果、アルツハイマー病の病期、フロルタウシピル(18F)を用いたPET検査を実施した場合はその結果等を考慮した上で、本剤投与の可否を判断すること。
【6.用法及び用量】(一部改訂)
通常、成人にはドナネマブ(遺伝子組換え)として初回は350mg、2回目は700mg、3回目は1050mg、その後は1回1400mgを4週間隔で、少なくとも30分かけて点滴静注する。
【8.重要な基本的注意】(一部改訂)
ARIAを示唆する症状が認められない場合であっても、本剤2回目の投与前、4回目の投与前、及び7回目の投与前、並びにそれ以降も定期的にMRI検査を実施し、ARIAの有無を確認すること。また、多くの重篤なARIAは治療開始12週以内にあらわれるので、必要に応じて本剤3回目の投与前にもMRI検査を実施することが望ましい。
【8.重要な基本的注意】(一部改訂)
アポリポ蛋白E対立遺伝子4(APOEε4)(ホモ接合型又はヘテロ接合型)キャリアの患者において、ARIA-E、ARIA-H、及び重篤なARIA-E及びARIA-Hがより高い頻度で認められている。なお、発現頻度は、APOEε4(ホモ接合型)キャリアで最も高く、次にAPOEε4(ヘテロ接合型)キャリア、APOEε4ノンキャリアの順で高かった。APOEε4保因状況にかかわらず、8.2.1~8.2.3項及び11.1.2項に規定のMRI検査を含むARIA管理を実施すること。アルツハイマー病による軽度認知障害及び軽度の認知症患者を対象とした本剤の国際共同第Ⅲ相試験(AACI試験)及び海外第Ⅲ相試験(AACQ試験)におけるAPOEε4ホモ接合型キャリアの割合はそれぞれ16.7%及び10.1%であった。
【8.重要な基本的注意】(一部改訂)
AACI試験におけるAPOEε4遺伝子型別のARIA発現頻度注1)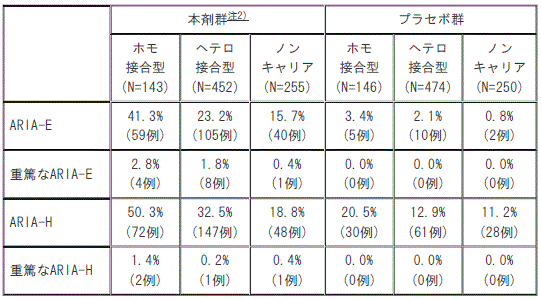
注1)MRI中央読影で認められたARIA及び治験担当医師により報告されたARIAから頻度を算出した。
注2)本剤を最初の3回は1回700mg、以降は1回1400mgを4週間隔で静脈内投与した(初回承認時の用法及び用量)。
【8.重要な基本的注意】(一部改訂)
AACQ試験におけるAPOEε4遺伝子型別のARIA発現頻度注3)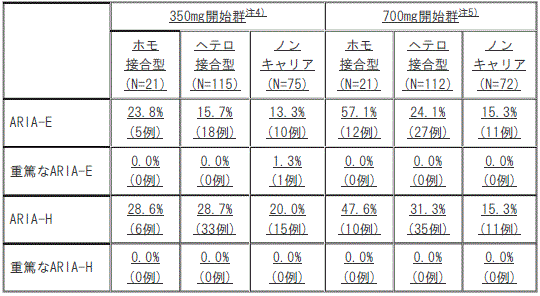
注3)MRI中央読影で認められたARIA及び治験担当医師により報告されたARIAから頻度を算出した。
注4)本剤を初回は350mg、2回目は700mg、3回目は1050mg、以降は1回1400mgを4週間隔で静脈内投与した。
注5)本剤を最初の3回は1回700mg、以降は1回1400mgを4週間隔で静脈内投与した(初回承認時の用法及び用量)。
【11.1重大な副作用】(一部改訂)
アミロイド関連画像異常(ARIA)、脳出血:
ARIA-E、ARIA-Hがあらわれることがある。また、重篤なARIAがあらわれることがあり、臨床試験において死亡に至った例が認められている。症候性ARIA-Eは2.8%で認められている。
(1)ARIAの症状としては、頭痛、錯乱、悪心、嘔吐、ふらつき、めまい、振戦、視覚障害、言語障害、認知機能の悪化、意識変容、発作等がある。ARIAを疑う症状が発現した場合にはMRI検査を実施すること。臨床試験で認められたARIA-Eの発現から消失までの中央値は約8週間であった。
(2)ARIA-Eについては、必要に応じてコルチコステロイド等による支持療法を行うこと。ARIA-Hの症状が認められた場合にはARIA-Eも併発していることが多いため、ARIA-E発現時と同様の処置を行うこと。
(3)ARIAは再発することがあるため、投与を再開した場合は、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討すること。
(4)ARIAが再発した患者において、本剤の投与を再開した経験は限られている。
【14.適用上の注意】(一部改訂)
[薬剤調製時の注意]
希釈液は、生理食塩液を用いること。下表に従い、本剤を必要量抜き取り、生理食塩液を含む点滴静注用バッグ又はボトルに添加して最終濃度が4~10mg/mLになるように希釈すること。
効能効果
アルツハイマー病による軽度認知障害及びアルツハイマー病による軽度の認知症の進行抑制。
(効能又は効果に関連する注意)
5.1. 本剤は、疾患の進行を完全に停止、又は疾患を治癒させるものではない。
5.2. 承認を受けた診断方法、例えばアミロイドPET、脳脊髄液(CSF)検査、又は同等の診断法によりアミロイドβ病理を示唆する所見が確認され、アルツハイマー病と診断された患者のみに本剤を使用すること。
5.3. 無症候でアミロイドβ病理を示唆する所見のみが確認できた者、及び中等度以降のアルツハイマー病による認知症患者に本剤を投与開始しないこと。
5.4. 「17.臨床成績」の項の内容を熟知し、第3相試験で用いられた診断基準、組み入れられた患者の臨床症状スコアの範囲、試験結果等を十分に理解した上で本剤投与の適否を判断すること〔17.1.1、17.1.2参照〕。
5.5. フロルタウシピル(18F)を用いたPET検査の結果から軽度以上のタウ蓄積が認められると判断できない患者に対する有効性及び安全性は確立していない。本剤の投与に先立ち、アミロイドβ病理に関する検査結果、アルツハイマー病の病期、フロルタウシピル(18F)を用いたPET検査を実施した場合はその結果等を考慮した上で、本剤投与の可否を判断すること。
用法用量
通常、成人にはドナネマブ(遺伝子組換え)として初回は350mg、2回目は700mg、3回目は1050mg、その後は1回1400mgを4週間隔で、少なくとも30分かけて点滴静注する。
(用法及び用量に関連する注意)
7.1. 安全性上の理由等で本剤1400mgに増量できない場合は、漫然と投与を継続しないこと。
7.2. 本剤投与中にアミロイドβプラークの除去が確認された場合は、その時点で本剤の投与を完了すること。アミロイドβプラークの除去が確認されない場合であっても、本剤の投与は原則として最長18ヵ月で完了すること。18ヵ月を超えて投与する場合は、18ヵ月時点までの副作用の発現状況、臨床症状の推移やアミロイドβプラークの変化等を考慮し、慎重に判断すること〔7.4参照〕。
7.3. アミロイドβプラークの除去は、アミロイドPET検査又は同等の診断法により評価し、検査を実施する場合の時期は本剤投与開始後12ヵ月を目安とすること。
7.4. 本剤投与中は6ヵ月毎を目安に認知機能検査、患者及び家族・介護者から自他覚症状の聴取等による臨床症状の評価を行い、臨床症状の経過、認知症の重症度等から本剤の有効性が期待できないと考えられる場合は本剤の投与を中止すること。なお、本剤投与中に認知症の重症度が中等度以降に進行した患者に投与を継続したときの有効性は確立していない〔7.2、17.1.1参照〕。
7.5. 本剤投与により、アミロイド関連画像異常(ARIA)としてARIA-浮腫/ARIA-滲出液貯留(ARIA-E)もしくはARIA-脳微小出血・ARIA-脳表ヘモジデリン沈着症(ARIA-H)、又は脳出血があらわれることがある〔1.2、2.2、2.3、8.2、8.2.2-8.2.4、11.1.2参照〕。
(1). ARIA-E
MRI画像上重度又は症候性のARIA-Eが認められた場合には、本剤の投与を中断又は中止すること。MRI画像上中等度かつ無症候性のARIA-Eが認められた場合には、本剤の投与を中断すること。MRI画像上軽度かつ無症候性のARIA-Eが認められた場合には、慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること。
本剤を中断し、ARIAの症状の消失及びMRI検査でのARIA-Eの消失を確認した場合には、投与の再開を検討することができる。
(2). ARIA-H及び脳出血
MRI画像上重度又は症候性のARIA-Hが認められた場合には、本剤の投与を中断又は中止すること。MRI画像上中等度かつ無症候性のARIA-Hが認められた場合には、本剤の投与を中断すること。MRI画像上軽度かつ無症候性のARIA-Hが認められた場合には、慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること。
本剤を中断し、ARIAの症状の消失及びMRI検査でのARIA-Hの安定化を確認した場合には、投与の再開を検討することができる。
1cmを超える脳出血が認められた場合には、本剤の投与を中止すること。
【参考】
1). ARIA-E(MRI所見)
①. 軽度:脳溝、皮質、又は皮質下白質の1ヵ所に限局した、5cm未満のFluid Attenuated Inversion Recovery(FLAIR)高信号。
②. 中等度:最大径が5~10cmのFLAIR高信号が1ヵ所にみられる、又は10cm未満の高信号が複数部位にみられる。
③. 重度:10cmを超えるFLAIR高信号で、脳回腫脹及び脳溝消失を伴う。1ヵ所又は複数ヵ所に独立した病変を認める。
2). ARIA-H(MRI所見)
①. 軽度:(脳微小出血)新規が1~4個、(脳表ヘモジデリン沈着症)1ヵ所。
②. 中等度:(脳微小出血)新規が5~9個、(脳表ヘモジデリン沈着症)2ヵ所。
③. 重度:(脳微小出血)新規が10個以上、(脳表ヘモジデリン沈着症)3ヵ所以上。
1). ARIA-E
①. 無症候性
a. 軽度無症候性ARIA-E:投与継続可能[慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること]。
b. 中等度無症候性ARIA-E:画像所見消失まで投与中断[MRI検査でのARIA-Eの消失を確認した場合には、投与の再開を検討することができる]。
c. 重度無症候性ARIA-E:画像所見消失まで投与中断[MRI検査でのARIA-Eの消失を確認した場合には、投与の再開を検討することができる]又は中止。
②. 症候性
軽度、中等度、重度症候性ARIA-E:症状及び画像所見消失まで投与中断[ARIAの症状の消失及びMRI検査でのARIA-Eの消失を確認した場合には、投与の再開を検討することができる]又は中止。
2). ARIA-H及び脳出血
①. 無症候性
a. 軽度無症候性ARIA-H:投与継続可能[慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること]。
b. 中等度無症候性ARIA-H:画像所見安定化まで投与中断[MRI検査でのARIA-Hの安定化を確認した場合には、投与の再開を検討することができる]。
c. 重度無症候性ARIA-H:画像所見安定化まで投与中断[MRI検査でのARIA-Hの安定化を確認した場合には、投与の再開を検討することができる]又は中止。
d. 画像上1cmを超える無症候性脳出血:投与中止。
②. 症候性
a. 軽度、中等度、重度症候性ARIA-H:症状消失及び画像所見安定化まで投与中断[ARIAの症状の消失及びMRI検査でのARIA-Hの安定化を確認した場合には、投与の再開を検討することができる]又は中止。
b. 画像上1cmを超える症候性脳出血:投与中止。
1). ARIA-E
①. 軽度:ARIA重症化の有無を確認するため、軽度ARIA-Eが無症候性で投与を継続する場合、発現から約1~2ヵ月後にMRI検査の実施を考慮する。軽度ARIA-Eが無症候性で投与を中断する場合、又は症候性の場合は、発現から約2~4ヵ月後にMRI検査を実施し、画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施する。
②. 中等度、重度:中等度、重度ARIA-E発現から約2~4ヵ月後にMRI検査を実施し、画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施する。
2). ARIA-H及び脳出血
①. 軽度:軽度ARIA-Hが症候性の場合、発現から約2~4ヵ月後にMRI検査を実施し、画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施する。
②. 中等度、重度:中等度、重度ARIA-H発現から約2~4ヵ月後にMRI検査を実施し、画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施する。
③. 1cmを超える脳出血:画像上1cmを超える脳出血発現後、臨床評価に基づき適宜MRI検査を実施する。
警告・禁忌・相互作用・その他の注意
(警告)
1.1. 本剤の投与は、アミロイドPET、MRI等の本剤投与にあたり必要な検査及び管理が実施可能な医療施設又は当該医療施設と連携可能な医療施設において、アルツハイマー病の病態、診断、治療に関する十分な知識及び経験を有し、本剤のリスク等について十分に管理・説明できる医師の下で、本剤の投与が適切と判断される患者のみに投与を行うこと。
1.2. 本剤の投与開始に先立ち、本剤投与によるARIAの発現割合、ARIAのリスク及びリスク管理のために必要な検査、ARIA発現時の対処法について患者及び家族・介護者に十分な情報を提供して説明し、同意を得てから投与すること。また、異常が認められた場合には、速やかに主治医に連絡するよう指導すること〔7.5、8.2、8.2.1-8.2.4、11.1.2参照〕。
(禁忌)
2.1. 本剤の成分に対し重篤な過敏症の既往歴のある患者。
2.2. 本剤投与開始前に血管原性脳浮腫が確認された患者[ARIAのリスクが高まるおそれがある]〔7.5、8.2.1参照〕。
2.3. 本剤投与開始前に5個以上の脳微小出血、投与開始前に脳表ヘモジデリン沈着症又は投与開始前に1cmを超える脳出血が確認された患者[ARIAのリスクが高まるおそれがある]〔7.5、8.2.1参照〕。
(重要な基本的注意)
8.1. アナフィラキシーを含むinfusion reactionがあらわれることがあるため、本剤投与終了後少なくとも30分は患者の状態を観察すること〔11.1.1参照〕。
8.2. 本剤はARIA管理に関する適切な知識を有する医師の下で使用し、投与開始前及び投与中は次の点に注意すること〔1.2、7.5、11.1.2参照〕。
8.2.1. 本剤投与開始前に、最新(1年以内)のMRI画像により、ARIAを含む異常所見の有無を確認すること〔1.2、2.2、2.3、8.2.4、8.3、11.1.2、17.1.1参照〕。
8.2.2. ARIAの発現は、本剤投与開始から24週間以内に多く、重篤なARIAの発現は投与開始から12週間以内に多いことから、この期間は特に注意深く患者の状態を観察すること。当該期間にかかわらず、ARIAを示唆する症状(頭痛、錯乱、悪心、嘔吐、ふらつき、めまい、振戦、視覚障害、言語障害、認知機能の悪化、意識変容、発作等)が認められた場合には、速やかに医療機関を受診するように患者及び家族・介護者に指導すること。速やかに臨床評価を行い、ARIAの発現が疑われる場合はMRI検査を実施すること〔1.2、7.5、8.2.4、11.1.2参照〕。
8.2.3. ARIAを示唆する症状が認められない場合であっても、本剤2回目の投与前、3回目の投与前、4回目の投与前、及び7回目の投与前、並びにそれ以降も定期的にMRI検査を実施し、ARIAの有無を確認すること〔1.2、7.5、8.2.4、11.1.2参照〕。
8.2.4. アポリポ蛋白E対立遺伝子4
[AACI試験におけるAPOEε4遺伝子型別のARIA発現頻度*]
1). ARIA-E
①. 本剤群※:ホモ接合型(N=143)41.3%(59例)、ヘテロ接合型(N=452)23.2%(105例)、ノンキャリア(N=255)15.7%(40例)。
②. プラセボ群:ホモ接合型(N=146)3.4%(5例)、ヘテロ接合型(N=474)2.1%(10例)、ノンキャリア(N=250)0.8%(2例)。
2). 重篤なARIA-E
①. 本剤群※:ホモ接合型(N=143)2.8%(4例)、ヘテロ接合型(N=452)1.8%(8例)、ノンキャリア(N=255)0.4%(1例)。
②. プラセボ群:ホモ接合型(N=146)0.0%(0例)、ヘテロ接合型(N=474)0.0%(0例)、ノンキャリア(N=250)0.0%(0例)。
3). ARIA-H
①. 本剤群※:ホモ接合型(N=143)50.3%(72例)、ヘテロ接合型(N=452)32.5%(147例)、ノンキャリア(N=255)18.8%(48例)。
②. プラセボ群:ホモ接合型(N=146)20.5%(30例)、ヘテロ接合型(N=474)12.9%(61例)、ノンキャリア(N=250)11.2%(28例)。
4). 重篤なARIA-H
①. 本剤群※:ホモ接合型(N=143)1.4%(2例)、ヘテロ接合型(N=452)0.2%(1例)、ノンキャリア(N=255)0.4%(1例)。
②. プラセボ群:ホモ接合型(N=146)0.0%(0例)、ヘテロ接合型(N=474)0.0%(0例)、ノンキャリア(N=250)0.0%(0例)。
*)MRI中央読影で認められたARIA及び治験担当医師により報告されたARIAから頻度を算出した。
※)本剤を最初の3回は1回700mg、以降は1回1400mgを4週間隔で静脈内投与した(初回承認時の用法及び用量)。
[AACQ試験におけるAPOEε4遺伝子型別のARIA発現頻度*]
1). ARIA-E
①. 350mg開始群※:ホモ接合型(N=21)23.8%(5例)、ヘテロ接合型(N=115)15.7%(18例)、ノンキャリア(N=75)13.3%(10例)。
②. 700mg開始群※※:ホモ接合型(N=21)57.1%(12例)、ヘテロ接合型(N=112)24.1%(27例)、ノンキャリア(N=72)15.3%(11例)。
2). 重篤なARIA-E
①. 350mg開始群※:ホモ接合型(N=21)0.0%(0例)、ヘテロ接合型(N=115)0.0%(0例)、ノンキャリア(N=75)1.3%(1例)。
②. 700mg開始群※※:ホモ接合型(N=21)0.0%(0例)、ヘテロ接合型(N=112)0.0%(0例)、ノンキャリア(N=72)0.0%(0例)。
3). ARIA-H
①. 350mg開始群※:ホモ接合型(N=21)28.6%(6例)、ヘテロ接合型(N=115)28.7%(33例)、ノンキャリア(N=75)20.0%(15例)。
②. 700mg開始群※※:ホモ接合型(N=21)47.6%(10例)、ヘテロ接合型(N=112)31.3%(35例)、ノンキャリア(N=72)15.3%(11例)。
4). 重篤なARIA-H
①. 350mg開始群※:ホモ接合型(N=21)0.0%(0例)、ヘテロ接合型(N=115)0.0%(0例)、ノンキャリア(N=75)0.0%(0例)。
②. 700mg開始群※※:ホモ接合型(N=21)0.0%(0例)、ヘテロ接合型(N=112)0.0%(0例)、ノンキャリア(N=72)0.0%(0例)。
*)MRI中央読影で認められたARIA及び治験担当医師により報告されたARIAから頻度を算出した。
※)本剤を初回は350mg、2回目は700mg、3回目は1050mg、以降は1回1400mgを4週間隔で静脈内投与した。
※※)本剤を最初の3回は1回700mg、以降は1回1400mgを4週間隔で静脈内投与した(初回承認時の用法及び用量)。
8.3. 本剤投与開始前のMRI検査で重度白質病変が認められた患者において、本剤の投与を開始した経験はない(重度の白質病変が認められた患者への本剤投与の可否は、本剤投与によるリスクとベネフィットを考慮した上で、慎重に判断すること)〔8.2.1参照〕。
8.4. 一般的に高血圧症は脳出血のリスク因子であることから、本剤投与前に高血圧の有無を確認し、高血圧が持続する患者への投与は慎重に行うこと(本剤投与中は適切な血圧管理を行うこと)。
(特定の背景を有する患者に関する注意)
(妊婦)
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること(生殖発生毒性試験は実施していない、また、一般にヒトIgGは胎盤を通過することが知られている)。
(授乳婦)
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること(本剤の乳汁中への移行に関するデータはないが、ヒトIgGは乳汁中へ移行することが知られている)。
(相互作用)
10.2. 併用注意:
1). 血液凝固阻止剤(ワルファリンカリウム、ヘパリンナトリウム、アピキサバン等)、血小板凝集抑制作用を有する薬剤(アスピリン、クロピドグレル硫酸塩等)[本剤との併用によりARIA-H又は脳出血が起こる可能性があるので、併用時にはARIA-H及び脳出血に注意すること(本剤の副作用としてARIA-Hの報告があり、併用によりこれらの薬剤が出血を助長する可能性がある)]。
2). 血栓溶解剤(アルテプラーゼ等)[本剤との併用によりARIA-H又は脳出血が起こる可能性があるので、併用時にはARIA-H及び脳出血に注意し、ARIAと虚血性脳卒中は類似した局所神経脱落症候を呈する場合があるので、本剤投与中の患者で虚血性脳卒中に対する血栓溶解療法を行う前に、これらの症候がARIAによるものである可能性を考慮すること(本剤の副作用としてARIA-Hの報告があり、併用によりこれらの薬剤が出血を助長する可能性がある)]。
(適用上の注意)
14.1. 薬剤調製時の注意
14.1.1. 本剤は1回使い切りのバイアル製剤である。本剤は、無菌的に希釈調製を行うこと。
14.1.2. 調製の約30分前に冷蔵庫から取り出し、室温に戻しておくこと。
14.1.3. バイアル内の薬液に異物や変色が認められないことを確認し、異物や変色が認められる場合は使用しないこと。
14.1.4. 希釈液は、生理食塩液を用いること。次に従い、本剤を必要量抜き取り、生理食塩液を含む点滴静注用バッグ又はボトルに添加して最終濃度が4~10mg/mLになるように希釈すること。
1). 投与量350mg(本剤1バイアル、20mL):生理食塩液の量15~67.5mL。
2). 投与量700mg(本剤2バイアル、合計40mL):生理食塩液の量30~135mL。
3). 投与量1050mg(本剤3バイアル、合計60mL):生理食塩液の量45~202.5mL。
4). 投与量1400mg(本剤4バイアル、合計80mL):生理食塩液の量60~270mL。
14.1.5. 点滴静注用バッグ又はボトルの中身をゆっくり反転させて混和し、激しく振とうしないこと。
14.1.6. 調製後は、速やかに使用すること(なお、やむを得ず保存を必要とする場合は、凍結を避け、冷蔵保存(2~8℃)では72時間以内、25℃以下での保存では12時間以内に使用すること)。
14.2. 薬剤投与時の注意
本剤投与終了後は、点滴ラインを生理食塩液にてフラッシュし、全量を投与すること。
(その他の注意)
15.1. 臨床使用に基づく情報
臨床試験において、本剤を投与された88%の患者で抗薬物抗体(ADA)が認められ、その全例で中和抗体が認められた。ADA陽性例ではADA陰性例と比較して血清中ドナネマブ濃度が低下する傾向が認められたが、ADAの発現による本剤の有効性への明らかな影響は認められなかった。注入に伴う反応が認められたすべての患者でADAが認められた。
(取扱い上の注意)
20.1. 外箱開封後は遮光して保存すること。
20.2. 凍結を避けること。凍結した場合は使用しないこと。
20.3. 激しく振とうしないこと。
20.4. 冷蔵庫(2~8℃)で保存できない場合は25℃以下で遮光保存し、3日以内に使用すること。
(参考情報:「7.用法及び用量に関連する注意」の補足情報)
1). ARIA-Eの発現
①. ARIA-Eの発現→無症候性→画像上軽度→投与継続*→ARIA重症化有無確認のため、発現から約1~2ヵ月後にMRI検査の実施を考慮。また、8.2.2/8.2.3項の残りのMRI検査に従う。
②. ARIA-Eの発現→無症候性→画像上中等度→投与中断→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
③. ARIA-Eの発現→無症候性→画像上中等度→投与中断→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与中止。
④. ARIA-Eの発現→無症候性→画像上重度→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
⑤. ARIA-Eの発現→無症候性→画像上重度→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与中止。
⑥. ARIA-Eの発現→症候性→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
⑦. ARIA-Eの発現→症候性→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施→症状及び画像所見の消失※※→投与中止。
*)慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること。無症候性で投与を中断する場合は、中等度、重度のMRIモニタリングに準ずる。
※)投与を中止した場合、発現から約2~4ヵ月後にMRI検査を実施する。画像上ARIA-Eの消失が確認されない場合は、追加のMRI検査を実施する。
※※)症状及び画像所見の消失を確認した場合、投与の再開を検討することができる。
2). ARIA-Hの発現
①. ARIA-Hの発現→無症候性→画像上軽度→投与継続*→8.2.2/8.2.3項の残りのMRI検査に従う。
②. ARIA-Hの発現→無症候性→画像上中等度→投与中断→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
③. ARIA-Hの発現→無症候性→画像上中等度→投与中断→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与中止。
④. ARIA-Hの発現→無症候性→画像上重度→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
⑤. ARIA-Hの発現→無症候性→画像上重度→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与中止。
⑥. ARIA-Hの発現→症候性→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与再開→ARIAは再発することがあるため、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討。また、8.2.3項の残りのMRI検査に従う。
⑦. ARIA-Hの発現→症候性→投与中断又は中止※→発現から約2~4ヵ月後にMRI検査を実施。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施→症状消失及び画像所見の安定化※※→投与中止。
3). 画像上1cmを超える脳出血→投与中止→臨床評価に基づき適宜MRI検査を実施。
*)慎重に臨床評価した上で、本剤の投与継続の可否を検討し、投与継続する場合、特に注意深く経過観察すること。
※)投与を中止した場合、発現から約2~4ヵ月後にMRI検査を実施する。画像上ARIA-Hの安定化が確認されない場合は、追加のMRI検査を実施する。
※※)症状消失及び画像所見の安定化を確認した場合、投与の再開を検討することができる。
(保管上の注意)
2~8℃で冷蔵保存。
副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1. 重大な副作用
11.1.1. Infusion reaction(17.9%*)、アナフィラキシー(0.5%*):アナフィラキシーを含むinfusion reaction(紅斑、悪寒、悪心、嘔吐、発汗、頭痛、胸部絞扼感、呼吸困難、血圧変動等)があらわれることがあり、重症又は致命的な経過をたどるおそれがある(多くは本剤投与中又は投与終了後30分以内に発現する)〔8.1参照〕。
11.1.2. アミロイド関連画像異常(ARIA)(28.8%*※)、脳出血(0.5%*※):ARIA-E(15.6%*※)、ARIA-H(25.5%*※)があらわれることがある。また、重篤なARIA(0.5%*※)があらわれることがあり、臨床試験において死亡に至った例が認められている。症候性ARIA-Eは2.8%*※で認められている〔1.2、7.5、8.2、8.2.1-8.2.4参照〕。
(1). ARIAの症状としては、頭痛、錯乱、悪心、嘔吐、ふらつき、めまい、振戦、視覚障害、言語障害、認知機能悪化、意識変容、発作等がある(ARIAを疑う症状が発現した場合にはMRI検査を実施すること)。臨床試験で認められたARIA-Eの発現から消失までの中央値は約8週間であった。
(2). ARIA-Eについては、必要に応じてコルチコステロイド等による支持療法を行うこと。ARIA-Hの症状が認められた場合にはARIA-Eも併発していることが多いため、ARIA-E発現時と同様、必要に応じてコルチコステロイド等による支持療法を行うこと。
(3). ARIAは再発することがあるため、投与を再開した場合は、注意深く患者の状態を観察するとともに、定期的なMRI検査の実施を検討すること。
(4). ARIA再発した患者において、本剤の投与を再開した経験は限られている。
11.2. その他の副作用
1). 胃腸障害:(1%以上)*悪心、(頻度不明)嘔吐。
2). 神経系障害:(1%以上)*頭痛。
*)AACQ試験における発現頻度の集計に基づき記載した(本剤を初回は350mg、2回目は700mg、3回目は1050mg、以降は1回1400mgを4週間隔で静脈内投与した)。
※)MRI中央読影で認められたARIA又は脳出血及び治験担当医師により報告されたARIA又は脳出血から頻度を算出した。
薬物動態
16.1 血中濃度
16.1.1 単回投与
アルツハイマー病による軽度認知障害患者又はアルツハイマー病による軽度から中等度の認知症患者18例(日本人5例を含む)にドナネマブ10、20、及び40mg/kg注)(体重70kgの場合それぞれ700、1400、及び2800mg注))、並びにアルツハイマー病による軽度認知障害患者及び軽度の認知症患者(外国人212例)に350mgを単回静脈内投与したときのドナネマブの曝露量はおおむね用量に比例して上昇した。消失半減期は約8~10日であった。
表1)アルツハイマー病による軽度認知障害患者又はアルツハイマー病による軽度から中等度の認知症患者にドナネマブ10、20、及び40mg/kg注)を単回静脈内投与したときのドナネマブの薬物動態パラメータ
<<表省略>>
図1)アルツハイマー病による軽度認知障害患者又はアルツハイマー病による軽度から中等度の認知症患者にドナネマブ10、20、及び40mg/kg注)を単回静脈内投与したときの血清中ドナネマブ濃度推移(平均値±標準偏差)
注)本剤の承認された用法及び用量は、「通常、成人にはドナネマブ(遺伝子組換え)として1回700mgを4週間隔で3回、その後は1回1400mgを4週間隔で、少なくとも30分かけて点滴静注する。」である。
<<図省略>>
16.1.2 反復投与
第I相~第III相試験のデータを用いて母集団薬物動態解析を実施した。ADAが陰性の患者に本剤1400mg注)を4週間ごとに反復静脈内投与したときの定常状態における薬物動態パラメータ[中央値(90%信頼区間)]は、Cmax,ssが381μg/mL(255、559)、Cmin,ssが22.2μg/mL(5.63、55.3)、AUCτ,ssが53500μg・h/mL(34900、91500)、と推定された。
16.3 分布
ドナネマブは静脈内投与後二相性で消失する。母集団薬物動態解析により推定された中央コンパートメントの分布容積は3.36L(個体間変動18.7%)、末梢コンパートメントの分布容積は4.83L(個体間変動93.9%)であった。
16.4 代謝
ドナネマブはIgG1モノクローナル抗体であり、内因性IgGと同様に異化経路によりペプチド断片及びアミノ酸に分解され、代謝酵素の阻害や誘導はないと考えられる。ドナネマブはチトクロムP450等の代謝酵素による代謝を受けないため、活性代謝物はない。
16.5 排泄
母集団薬物動態解析より推定されたクリアランスは0.0255L/h(個体間変動24.9%)、消失半減期は12.1日であった。
16.6 特定の背景を有する患者
母集団薬物動態解析によると、年齢、性別、人種、腎機能、及び肝機能はドナネマブの薬物動態に影響を及ぼさなかった。
注)本剤の承認された用法及び用量は、「通常、成人にはドナネマブ(遺伝子組換え)として初回は350mg、2回目は700mg、3回目は1050mg、その後は1回1400mgを4週間隔で、少なくとも30分かけて点滴静注する。」である。
臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験(AACI試験)
アルツハイマー病による軽度認知障害及び軽度の認知症患者1736例(日本人88例を含む)を対象としたプラセボ対照二重盲検比較試験を実施した。プラセボ又は本剤(最初の3回は1回700mg、以降は1回1400mg)を4週間隔で静脈内投与注1)した。主な選択基準は、①~④のとおりであった。
①MMSE注2)スコアが20~28である。
②アミロイドPET検査でアミロイドβプラーク沈着が認められる。
③フロルタウシピル(18F)を用いたPET検査の視覚読影と定量解析を組み合わせた判定において軽度以上のタウ蓄積が認められる。
④スクリーニング期の脳MRI検査で、次に示すような所見が認められていない。
・ARIA-E
・5ヵ所以上の脳微小出血
・2ヵ所以上の脳表ヘモジデリン沈着
・脳出血又は重度の白質病変
AACI試験では、事前に規定したPET検査でのアミロイドβプラーク除去の基準に基づき、二重盲検下にて本剤の投与を完了した。
76週時のiADRS注3)のベースラインからの変化量(主要評価項目)で評価した臨床的進行は、プラセボ群と比べて本剤群で統計学的に有意に抑制されており、抑制率は22.3%[タウ蓄積が軽度から中等度(SUVr≦1.46)及び高度(SUVr>1.46)の全体集団(以降、全体集団)]及び35.1%(タウ蓄積が軽度から中等度の集団)であった。ベースラインから投与後76週までの有効性の結果及びアミロイドPETセンチロイド値を表1、APOEε4遺伝子型別の有効性の結果を表2に示す。52週及び76週時に本剤群でアミロイドPET検査での陰性に相当するアミロイドβプラーク除去を達成した割合はそれぞれ66.1%及び76.4%で、52週及び76週時に本剤の投与完了基準を満たした割合はそれぞれ46.6%及び69.2%であった。
注1)初回承認時の用法及び用量
注2)MMSE(Mini-Mental State Examination)は、認知機能を評価する簡易尺度。合計スコアは0から30の範囲をとり、スコア低値は障害の程度がより大きいことを示す。
注3)iADRS(integrated Alzheimer’s Disease Rating Scale)は、ADCS-iADL注4)及びADAS-Cog13注5)から算出される認知機能及び日常生活機能を評価する尺度。合計スコアは0から144の範囲をとり、スコア低値は障害の程度がより大きいことを示す。
注4)ADCS-iADL(Alzheimer’s Disease Cooperative Study-Activities of Daily Living Inventory、instrumental items)は、手段的日常生活動作(電話、買い物、家事、趣味、外出等)を評価する尺度。合計スコアは0から59の範囲をとり、スコア低値は障害の程度がより大きいことを示す。
注5)ADAS-Cog13(Alzheimer’s Disease Assessment Scale-13-item Cognitive subscale)は、アルツハイマー病で最も典型的に障害を受ける認知機能領域の評価。合計スコアは0から85の範囲をとり、スコア高値は障害の程度がより大きいことを示す。
図1)iADRSの経時的変化(最小二乗平均値±標準誤差)(全体集団)
<<図省略>>
図1)iADRSの経時的変化(最小二乗平均値±標準誤差)(タウ蓄積が軽度から中等度の集団)
<<図省略>>
表1)ベースラインから投与後76週までの有効性の結果及びアミロイドPETセンチロイド値(EES集団)
<<表省略>>
表2)APOEε4遺伝子型別の有効性の結果(EES集団、投与76週時)
<<表省略>>
各投与群における有害事象、重篤な有害事象、治験薬投与中止に至った有害事象の発現頻度は表3)のとおりであった。主な副作用は、本剤群853例において、ARIA-E(アミロイド関連画像異常-浮腫/滲出液貯留)23.8%(203例)注6)、ARIA-H(アミロイド関連画像異常-微小出血およびヘモジデリン沈着)19.0%(162例)注6)、注入に伴う反応8.3%(71例)、脳表ヘモジデリン沈着症6.0%(51例)、頭痛5.3%(45例)、脳微小出血2.1%(18例)であり、これらの事象についてプラセボ群874例では、ARIA-E1.9%(17例)注6)、ARIA-H6.8%(59例)注6)、注入に伴う反応0.5%(4例)、脳表ヘモジデリン沈着症1.0%(9例)、頭痛3.0%(26例)、脳微小出血1.1%(10例)であった。[5.4、7.4、8.2.1参照]
注6)治験担当医師により報告されたARIAに基づき算出した。MRI中央読影のみでARIAが認められた症例は含まない。
表3)有害事象の発現頻度
<<表省略>>
17.1.2 海外第III相試験(AACQ試験)
アミロイドβ病理を示唆する所見がアミロイドPET検査で認められたアルツハイマー病による軽度認知障害及び軽度の認知症患者843例を対象に複数の投与レジメンを用いて、ARIA-Eの発現への影響を評価する実薬対照二重盲検比較試験を実施した。本剤は投与レジメン1及び2を含めた複数のレジメンで投与した(表4)。
表4)投与レジメン
<<表省略>>
24週時のARIA-Eの発現頻度注7)(主要評価項目)は、350mg開始群で13.7%(29/212例)、700mg開始群で23.7%(49/207例)であった。
注7)MRI中央読影で認められたARIA及び治験担当医師により報告されたARIAから頻度を算出した。
24週時の各投与群における有害事象、重篤な有害事象、治験薬投与中止に至った有害事象の発現頻度は表5)のとおりであった。ARIA-Eを除く主な副作用は、350mg開始群212例において、注入に伴う反応17.0%(36例)、ARIA-H11.8%(25例)注8)、頭痛5.7%(12例)、脳表ヘモジデリン沈着症2.4%(5例)、浮動性めまい2.4%(5例)であり、これらの事象について700mg開始群207例では、注入に伴う反応13.5%(28例)、ARIA-H14.5%(30例)注8)、頭痛9.2%(19例)、脳表ヘモジデリン沈着症5.3%(11例)、浮動性めまい0.5%(1例)であった。
表5)24週時の有害事象の発現頻度
<<表省略>>
76週時の主な副作用は、350mg開始群212例において、注入に伴う反応17.9%(38例)、ARIA-H16.0%(34例)注8)、ARIA-E15.1%(32例)注8)、頭痛5.7%(12例)、脳表ヘモジデリン沈着症4.2%(9例)、浮動性めまい2.8%(6例)、脳微小出血2.4%(5例)であり、これらの事象について700mg開始群207例では、注入に伴う反応14.0%(29例)、ARIA-H18.8%(39例)注8)、ARIA-E23.2%(48例)注8)、頭痛10.1%(21例)、脳表ヘモジデリン沈着症6.3%(13例)、浮動性めまい1.0%(2例)、脳微小出血2.4%(5例)であった。[5.4参照]
注8)治験担当医師により報告されたARIAに基づき算出した。MRI中央読影のみでARIAが認められた症例は含まない。
薬効薬理
18.1 作用機序
ドナネマブは、脳内の不溶性アミロイドβプラークにのみ存在すると考えられるN3pG Aβ(N末端第3残基がピログルタミル化されたアミロイドβ)を標的とするヒト化IgG1モノクローナル抗体である。ドナネマブは、N3pG Aβに結合し、ミクログリアによる貪食作用を介したアミロイドβプラーク除去を促進すると考えられている。
18.2 臨床薬理作用
アルツハイマー病患者を対象とした臨床薬理試験において、ドナネマブは、アミロイドPETにより測定した脳内のアミロイドβプラークを減少させた。
類似した薬効の薬
よく一緒に見られている薬

医師の処方により使用する医薬品。
